


Abstract
The αvβ6 integrin is involved in the pathogenesis of cancer and fibrosis. A radiolabeled 20-amino-acid αvβ6-binding peptide, derived from the foot and mouth virus (NAVPNLRGDLQVLAQKVART [A20FMDV2]), has been developed to image αvβ6 levels preclinically. This study was designed to translate these findings into a clinical PET imaging protocol to measure the expression of αvβ6 in humans. Methods: Preclinical toxicology was undertaken, and a direct immunoassay was developed for 4-fluorobenzamide (FB)-A20FMDV2. Four healthy human subjects (2 male and 2 female) received a single microdose of 18F-FB-A20FMDV2 followed by a multibed PET scan of the whole body over more than 3 h. Results: There were no findings in the preclinical toxicology assessments, and no anti-A20FMDV2 antibodies were detected before or after dosing with the PET ligand. The mean and SD of the administered mass of 18F-FB-A20FMDV2 was 8.7 ± 4.4 μg (range, 2.7–13.0 μg). The mean administered activity was 124 ± 20 MBq (range, 98–145 MBq). There were no adverse or clinically detectable pharmacologic effects in any of the subjects. No significant changes in vital signs, laboratory study results, or electrocardiography results were observed. Uptake of radioactivity was observed in the thyroid, salivary glands, liver, stomach wall, spleen, kidneys, ureters, and bladder. Time–activity curves indicated that the highest activity was in the bladder content, followed by the kidneys, small intestine, stomach, liver, spleen, thyroid, and gallbladder. The largest component of the residence times was the voided urine, followed by muscle, bladder, and liver. Using the mean residence time over all subjects as input to OLINDA/EXM, the effective dose was determined to be 0.0217 mSv/MBq; using residence times from single subjects gave an SD of 0.0020 mSv/MBq from the mean. The critical organ was the urinary bladder, with an absorbed dose of 0.18 mGy/MBq. Conclusion: 18F-FB-A20FMDV2 successfully passed toxicology criteria, showed no adverse effects in this first-in-humans study, and has an effective dose that enables multiple scans in a single subject.
The αvβ6 integrin is a cell-surface adhesion receptor that, in its activated form, interacts with extracellular ligands bearing the arginine-glycine-aspartic acid (RGD) tripeptide sequence (1). It plays a role in the etiology and progression of several pathologic conditions, including cancer and fibrosis, and as such it is an important prognostic biomarker as well as a potential drug target.
Key ligands for αvβ6 include the latency-associated peptides (LAPs) of transforming growth factor (TGF) β1 and β3 (LAPβ1 and LAPβ3), as well as extracellular matrix ligands such as fibronectin, tenascin, and vitronectin (2,3). Binding of extracellular matrix ligands to the integrin receptor can promote cell adhesion, activation of intracellular signaling pathways, and local release of activated TGFβ from latent complexes in the matrix (1,4,5). αvβ6 is upregulated on many cancers, including pancreatic, breast, ovarian, colon, and over 90% of oral squamous cell carcinoma (6,7). Furthermore, expression of αvβ6 correlates with development of metastasis in gastric cancer (8), is linked with a dramatic reduction in survival from colon cancer (9), and is reported to promote both the survival and the invasive potential of carcinoma cells (10–13). αvβ6-mediated activation of TGFβ promotes myofibroblast differentiation, proliferation, and collagen synthesis as part of normal physiologic wound healing (14,15). However, when this process persists and fails to resolve, the result is a pathologic elaboration of extracellular matrix, which results in irreversible organ scarring, ultimately resulting in organ failure and death (16).
The foot and mouth disease virus (FMDV) uses αvβ6 to access the intracellular environment of the host and thus causes the symptoms of foot and mouth disease. In fact, the envelope protein of the virus contains a 20-amino-acid peptide sequence, NAVPNLRGDLQVLAQKVART (A20FMDV2) that mediates FMDV infection by binding to αvβ6 (17–19). Phage display analysis identified the DLXXL sequence as a key moiety responsible for αvβ6 specificity while having only minimal interactions with other RGD integrins (e.g., αvβ3, αvβ5, and aIIbβ3) (20). The high affinity and selectivity (21), as well as the automated and good-manufacturing-practice–compatible radiochemistry and preclinical radiodosimetry (M. Onega et al., unpublished data, 2017), of this A20FMDV2 peptide for αvβ6 have recently been described.
In this article, we describe the safety, tolerability, immunogenicity, biodistribution, and radiation dosimetry of 18F-4-fluorobenzamide (FB)-A20FMDV2 (also known as 18F-IMAFIB and 18F-GSK2634673) in preparation for its use as a PET ligand to delineate αvβ6 in humans. If successful, this PET ligand may be useful in the clinical management of patients with cancer or fibrosis, since levels of αvβ6 expression may be of prognostic value (22,23) and potentially useful for treatment decisions in clinical practice. In addition, 18F-FB-A20FMDV2 may prove valuable as an in vivo imaging tool to demonstrate target engagement during development of new therapies that target αvβ6. Before we began this first-in-humans study of 18F-FB-A20FMDV2, we identified various potential risks and developed strategies to manage them. Preclinical toxicology was conducted to support microdosing in humans (up to a maximum mass dose of 100 μg, as detailed in International Committee on Harmonization guideline M3[R2]). Potential for immunogenic risk was explored through development of an immunogenicity assay to monitor both preexisting antibodies and antibodies induced by exposure to this FMDV2 peptide within this study.
MATERIALS AND METHODS
Study Conduct
Subjects were screened and recruited at Hammersmith Medicines Research, London, U.K., and imaging assessments were conducted at the Imanova Centre for Imaging Sciences, London, U.K. The study was approved by the London–Brent Research Ethics Committee, U.K. (13/LO/1792), and permission to administer radioisotopes was obtained from the Administration of Radioactive Substances Advisory Committee of the U.K. (630/3925/30809). The PETAL study (PET Study of αvβ6 in Lungs) is listed on clinicaltrials.gov as “A Validation and Dosimetry Study of GSK2634673F PET Ligand” (NCT02052297 and RES116235), and the dosimetry data presented here form part of that study.
Subjects
Four healthy human subjects (2 male and 2 female; age range, 48–65 y) were enrolled in the study after providing written informed consent. The main inclusion criteria were an age of 45 y or older and no clinically significant illness or disease. The main exclusion criteria were prior exposure to more than 10 mSv of radiation over the past 3 y or more than 10 mSv in a single year, including the proposed study; previous or current exposure to animals that may harbor FMDV (FMDV2); and previous long-term (≥3 mo) residence in a country where FMDV2 is endemic (such as certain areas of Africa, Asia, and South America).
Preclinical Toxicology
FB-A20FMDV2 (a nonradioactive homolog of the proposed labeled PET peptide) was screened against a broad range of in vitro pharmacologic targets (receptors, ion channels, enzymes, and transporters) that were distinct from the intended therapeutic target (αvβ6 integrin receptor) to assess the potential for off-target interactions.
A limited number of nonclinical toxicity studies were performed to evaluate the potential toxicity of the triacetate salt of FB-A20FMDV2, based on the principles of the 2009 M3(R2) guideline of the International Committee on Harmonization to support the human microdose PET study. The hemolytic potential of FB-A20FMDV2 solutions in 0.9% w/v aqueous sodium chloride (Fresenius Kabi) at concentrations of 0.04 and 0.4 mg/mL were assessed in vitro in rat and human blood. These concentrations were selected to match the concentrations used in a subsequent rat toxicity study. Rats were selected as a relevant species because they are known to express αvβ6 integrin receptors (M. Onega et al., unpublished data, 2017; (24)). To examine the toxicity and irritancy potential, groups of 10 male and 10 female Crl:Wi (Han) rats (9–11 wk old) received a single intravenous dose of vehicle (0.9% w/v aqueous sodium chloride) or FB-A20FMDV2 at 0.2 mg/kg or 2 mg/kg, at a dose volume of 5 mL/kg. These doses were approximately 100-fold and 1,000-fold the maximum permissible human microdose on a mg/kg basis. The animals in this cohort were sacrificed 24 h after dosing. Additional groups of 5 male and 5 female rats were treated similarly and then remained off-dose for 14 d to investigate the regression or progression of any target organ toxicity present at the end of the treatment period or delayed onset of any target organ toxicity. All animal studies were ethically reviewed and performed in accordance with the Animals (Scientific Procedures) Act of 1986 and the GlaxoSmithKline Policy on the Care, Welfare, and Treatment of Laboratory Animals.
Immunogenicity Assay
A direct immunoassay was developed and validated using a pool of A20FMDV2 (GSK3225458) peptide labeled with biotin either at the N- or the C-terminus to capture antibodies to the peptide. Captured antibodies were detected using a horseradish peroxidase–conjugated polyclonal goat antihuman IgG (heavy and light chains [H+L]) reagent capable of binding to human antibodies regardless of isotype. A negative control was prepared from pooled normal human serum and was used to define sensitivity and to normalize the results. Screening and confirmation cut points were determined during method validation.
Serum samples for immunogenicity screening were obtained from all 4 subjects before administration of 18F-FB-A20FMDV2 and at follow-up (2–4 wk after administration of 18F-FB-A20FMDV2). Samples were stored at −70°C until assayed.
The materials used were as follows: A20FMDV2 peptide (GSK2634673E), N-terminal biotin-peptide (GSK3225458A), C-terminal [4-FBA]-peptide-biotin (GSK3228218A), SmartBlock (catalog no. 113-500; Candor), LowCross-Buffer (catalog no. 100-500; Candor), normal goat serum (catalog no. 7332500; Lampire), human healthy donor serum (Bioreclamation), biotinylated human IgG (catalog no. 009-060-003; Jackson), goat antihuman IgG (H+L) horseradish peroxidase conjugate (catalog no. 109-035-088; Jackson), goat anti-rabbit IgG (H+L) horseradish peroxidase conjugate (catalog no. 111-035-045; Jackson), streptavidin plate (catalog no. 436014; Nunc), 10× phosphate-buffered saline, 1% polysorbate 20 (GlaxoSmithKline Media Prep), rabbit anti-GSK2634673E positive control (PC) antibody pool (lot no. DIO2.3/197923; GlaxoSmithKline), human plasma fibronectin (lot no. 2361790; EMD Millipore), EXL-405 plate washer (Biotek), and SpectraMax plate reader (Molecular Devices).
Negative control was prepared by pooling 54 samples of normal human serum, and PC was prepared by spiking rabbit antipeptide antibody in negative control serum at 500 ng/mL for high PC and 25 ng/mL for low PC. Unlabeled peptide and N-/C-terminal biotin-labeled peptides (1:1) were tested for plate coating with different fractions of glycine eluded (PC1A, PC2A) and TEA eluded (high affinity and binding, PC1B, PC2B) from 2 batches of PCs at 1,000, 500, 100, and 10 ng/mL. Assay sensitivity, PC specificity, and RGD motif interference were evaluated during method validation.
The antipeptide antibody was detected by a validated direct binding enzyme-linked immunosorbent assay. Briefly, assay controls and samples were diluted at 1:50 in assay buffer containing biotin-peptides (2.5 μg/mL) and incubated for 3 h at room temperature. The preincubated samples were transferred to a streptavidin-coated plate for a 1-h incubation. After a wash step, a cocktail of horseradish peroxidase–conjugated goat antihuman IgG (H+L) and antirabbit IgG (H+L) was added to the plate and incubated for 1 h. After any unbound conjugate had been washed away, 3,3′,5,5′-tetramethylbenzidine substrate was added to the plate. The bound conjugates produced a color reaction, and the intensity of the signal was directly proportional to the amount of detected antibody in the sample. Biotinylated human IgGs were used as a plate PC (Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org).
To determine a screening cut point, 76 healthy subjects (38 male and 38 female) were tested 3 times by 2 analysts on 2 different days to determine potential positives (above the cut point) and negatives (below). For the confirmation assay, 20 randomly selected subjects (10 male and 10 female) were spiked with 25 ng/mL of PC, and the spiked (positive population) and unspiked (negative population) samples were then incubated in the presence and absence, respectively, of excess peptide (50 μg/mL).
Clinical serum samples (preadministration and at week 3) were collected from the 4 healthy subjects and tested in this immunogenicity assay.
Imaging
Healthy human subjects were imaged for up to 3.5 h on a Biograph 6 TruePoint PET/CT scanner (Siemens Healthcare) after intravenous administration of 18F-FB-A20FMDV2, prepared according to a previously published method (M. Onega et al., unpublished data, 2017). The effective dose for each attenuation-corrected CT scan in this study was estimated to be 2.3 mSv using the ImPACT CTDosimetry spreadsheet system (25).
Before injection, each subject underwent CT for attenuation correction over 6–7 PET bed lengths covering the vertex of the head to the mid thigh. CT scanning used a 130-kV tube potential and 15 effective mAs exposure, with a 0.6-s gantry rotation time and a spiral pitch of 1.5. CT image data were acquired with 6 × 3 mm channels and reconstructed to 5-mm slices at 3-mm intervals, with a 700-mm field of view.
For each subject, PET scanning was initiated at the time of injection, and a series of 6 whole-body scans was obtained. Each scan consisted of 6–7 bed positions (depending on subject size), with the duration of subsequent scans increasing from 1 min per bed position to 2, 5, 5, 7, and 7 min per bed position. Between whole-body scans 4 and 5, the subject left the scanner bed to void the bladder and to move about for around 20 min to limit discomfort. After this break, a second attenuation-corrected CT scan was obtained before PET imaging was resumed. PET images were reconstructed on a 256 × 256 matrix, with a zoom of 1.3 using ordered-subsets expectation-maximization with 3 iterations, 21 subsets, and a 5-mm gaussian image filter. Corrections for scatter, attenuation, and decay (to the start of each scan) were applied.
Image Analysis
Image analysis was performed on an Inveon Research Workplace workstation (version 4.0.1.14; Siemens Healthcare). Regions of interest (ROIs) were drawn on the PET/CT datasets, delineating as many organs relevant to dosimetry as possible, in particular those defined as source organs by OLINDA/EXM, version 1.1 (26). CT anatomy or PET uptake was used as a guide, with one set of ROIs being drawn for the first 4 scans and another set being drawn for scans 5 and 6, after the bladder void and rest. A separate bladder ROI was used for each frame to account for the change in size as the bladder filled and emptied over the course of the study. The activity concentrations in these ROIs were exported from Inveon Research Workplace and imported to Excel (Microsoft) for further analysis. Organ ROI activity concentrations were tabulated for each frame, to create integrated time–activity curves, with the activity in each organ at the end of the sixth scan being assumed to decay in situ without further redistribution. These values were then multiplied by the organ volume in the Cristy and Eckerman adult phantom (27) to calculate the organ residence time (the equivalent duration in hours that a single megabecquerel of activity is present in that organ per injected megabecquerel of radioligand [MBq⋅h/MBq]).
The bladder was treated separately, with the total activity in the bladder in each frame being input to a model using an exponential fill and void, fitting parameters for the exponential rate and magnitude, and the void time and voided fraction. This model was then used with a 2-h voiding period fitted to the measured data and integrated to calculate the bladder residence time.
The total residence times for all organs for an injected radioligand should add to the mean lifetime for that isotope, given by the half-life divided by the natural logarithm of 2, which for 18F is 2.64 h. A remainder residence time was assigned to the injected activity that was not measured within ROIs in the image, given by the mean lifetime minus the total residence time over all organs and voided urine.
The mean organ residence times over all subjects were used as the input to OLINDA/EXM, which calculated the absorbed dose to each of the relevant target organs (mGy/MBq) and provided the resulting effective dose per unit of injected activity (mSv/MBq). Organs without specific tissue weighting factors result in an effective dose contribution of 0, as do the testes (because OLINDA/EXM uses the higher of the testis and ovary absorbed doses to characterize the gonad dose).
To provide a measure of the variability of this result, the effective dose per unit injected activity was also calculated using the residence times from each individual subject.
RESULTS
Preclinical Toxicology
FB-A20FMDV2 did not demonstrate any off-target activity when screened against a broad range of in vitro pharmacologic targets (receptors, ion channels, enzymes, and transporters), including a cardiovascular liability panel. Since FB-A20FMDV2 is a large-molecular-weight peptide, access to the ion channels was limited and it was considered unlikely that the peptide would affect the hERG channel. Therefore, taken together, further nonclinical safety pharmacologic assessment was not considered necessary.
The hemolytic potential of FB-A20FMDV2 in 0.9% w/v aqueous sodium chloride at concentrations of 0.04 and 0.4 mg/mL was assessed in vitro in rat and human blood. As anticipated, no evidence of hemolysis was observed at either concentration tested, and therefore, this formulation was deemed suitable for intravenous use. Local irritancy and target organ toxicity were investigated in an intravenous extended single-dose toxicity study in rats at 0.2 or 2 mg/kg with a 14-d off-dose period. Both doses were well tolerated, and no treatment-related clinical signs of toxicity, changes in body weight gain, or food consumption were observed. In addition, no treatment-related changes in clinical pathology parameters (hematology, clinical chemistry, and coagulation) were observed at either dose. At both terminal necropsy and after the 14-d off-dose period, there were no FB-A20FMDV2–related macroscopic or microscopic observations. Stage-dependent qualitative evaluation of spermatogenesis in the testes was performed on all male rats given vehicle and a 2 mg/kg dose of FB-A20FMDV2. The testes revealed normal progression of the spermatogenic cycle, and the expected cell associations and proportions in the various stages of spermatogenesis were present.
Thus, 18F-FB-A20FMDV2 was shown to be nontoxic and to support microdosing in humans up to 100 μg (equivalent to 2 μg/kg for a 50-kg individual), with anticipated cover (at the human dose) of 1,000-fold on a mg/kg basis.
Clinical Safety and Tolerability
The PET scans were well tolerated, and all 4 subjects completed the scans as planned. None of the 4 subjects had any adverse events associated with the 18F-FB-A20FMDV2 or the PET procedure. Three adverse events occurred in 2 subjects: abdominal cramps in subject 4, erythema of the conjunctiva in subject 1, and heaviness of the arms in subject 4. Subject 4 had the lowest mass injected (2.7 μg), whereas subject 1 had the highest mass administered (13 μg). Therefore, there was no relationship to the mass administered. All adverse events were mild and transient and resolved the same day. Causality was determined by the study physicians based on their combined experience with phase I trials and with studies of other novel radiotracers. We believe that none of these adverse effects was due to the radioligand or the imaging procedure.
Immunogenicity
The screening and confirmation cut points were statistically established during validation and were 2.10 relative optical density (ROD) and 48.4% inhibition, respectively. To differentiate peptide-boosted true-positive from the preexisting positive responses in a healthy population, a ratio of postdose ROD over predose ROD was calculated. Samples with a ratio of 2 or more were reported as peptide-boosted positives. On the basis of clinical sample testing results, all 4 human subjects were negative in the antipeptide antibody assays.
To avoid epitope masking due to plate coating or biotinylation of peptide, a mixture of N- and C-terminal biotinylation peptides was tested on 96-well streptavidin plates, compared with unlabeled peptide on regular plates. The results showed that the signals of PC1A were higher than those of PC2A; PC2B and PC2B had very low or negative responses on peptide-coated plates. The sensitivity of the peptide-coated plate assay was poor (≥100 ng/mL). However, all PCs had strong and similar responses on biotin-peptide plates (Supplemental Figs. 3 and 4). Assay PC did not have a cross-reaction to endogenous RGD-containing proteins, and its signals were not inhibited in the presence of fibronectin at concentrations of up to 50 μg/mL (Supplemental Fig. 4). The percentage inhibition of PC at 20, 100, and 500 ng/mL by peptide were 53%, 73%, and 79%, respectively (Supplemental Fig. 5). The sensitivity was demonstrated to be 9.2 ng of PC per milliliter. The peptide interference level was 100 μg of peptide per milliliter at 100 ng of PC per milliliter. The intrarun and interrun precision for PC samples was 16.9% and 9.0%, respectively.
The screening and confirmation cut points were statistically established during validation and were 2.10 ROD and 48.41% inhibition, respectively. Among 75 healthy subjects, 17% (13/76) were screened and confirmed positive by peptide inhibition and excluded for screening cut point determination. To differentiate peptide-induced true-positives from the prevalence of positive responses in clinical subjects, a ratio of predose ROD over postdose ROD was calculated for all subjects who had a confirmed predose positive sample. Samples with a ratio of 2 or more were reported as peptide-induced positives. On the basis of clinical sample testing results, all 4 human subjects were negative in antipeptide antibody assays.
The A20FMDV2 peptide has 100% homology with FMDV, type O, VP1 139-158. The peptide strongly and specifically binds to αvβ6 receptor through its RGD motifs. Compared with integrin receptor ligands in vivo, the sequences 1–6 and 11–20 are nonhumanized and may potentially raise antibodies (Supplemental Fig. 5). The RGD motifs of peptide may also be immunogenic to humans (Supplemental Fig. 6). Furthermore, if a polyclonal PC contains anti-RGD motif antibodies, it may also bind to ligands such as fibronectin, tenascin-C, fibrinogen, LAP-TGFβ, and vitronectin in serum, resulting in an assay issue (the positive control may bind to peptides other than A20FMDV2 resulting in spurious data). Since fibronectin (molecular weight, 460 kDa; dimer) is one of the ligands that specifically binds to αv and β6 chains of the receptor, it was tested with PC during method development to evaluate PC specificity. The results showed that rabbit polyclonal antipeptide antibody did not bind to fibronectin or other RGD motif proteins in human serum and could be used as a PC for the antipeptide antibody assays.
Use of both N- and C-terminal labeled peptides as the capture reagents can potentially eliminate epitope-masking–induced false-negatives and enhance the direct enzyme-linked immunosorbent assay’s sensitivity and drug tolerance. This approach may be applied when immunogenicity methods are developed for other small-peptide biotherapeutics if there is concern about epitope-masking when labeling of the peptide is required or is directly immobilized on a solid surface. Furthermore, this direct enzyme-linked immunosorbent assay format can be used for screening, confirmation, titration, and isotyping assays. The antipeptide antibody method has been successfully used for this study of 4 healthy subjects and for other ongoing studies (data not shown) and will be further validated in patients with idiopathic pulmonary fibrosis. The impact of preexisting antibodies on efficacy, safety, and immunogenicity will be studied.
PET Imaging
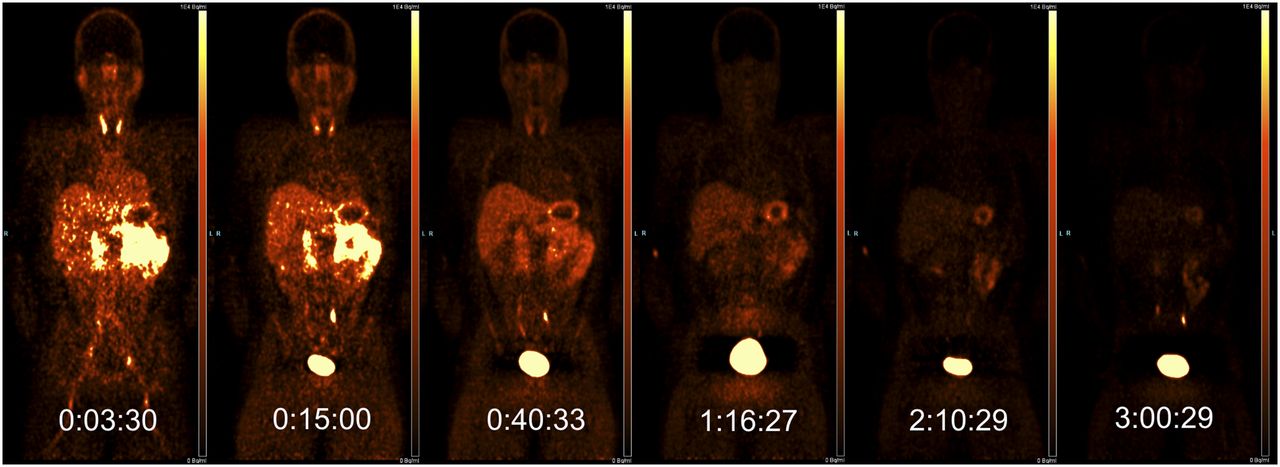
Four subjects were scanned, with demographic and radioligand administration details given in Table 1. PET images, through a single coronal slice, are presented in Figure 1 and show the activity uptake and clearance by tissue organs over the more than 3-h total study time for subject 1. Organs that are clearly visible in this slice through the body include the thyroid, salivary glands, liver, stomach wall, spleen, ureters, and bladder. Figure 2 shows coronal and sagittal CT views through subject 1, overlaid with organ ROIs.
Subject Demographic and Scan Details
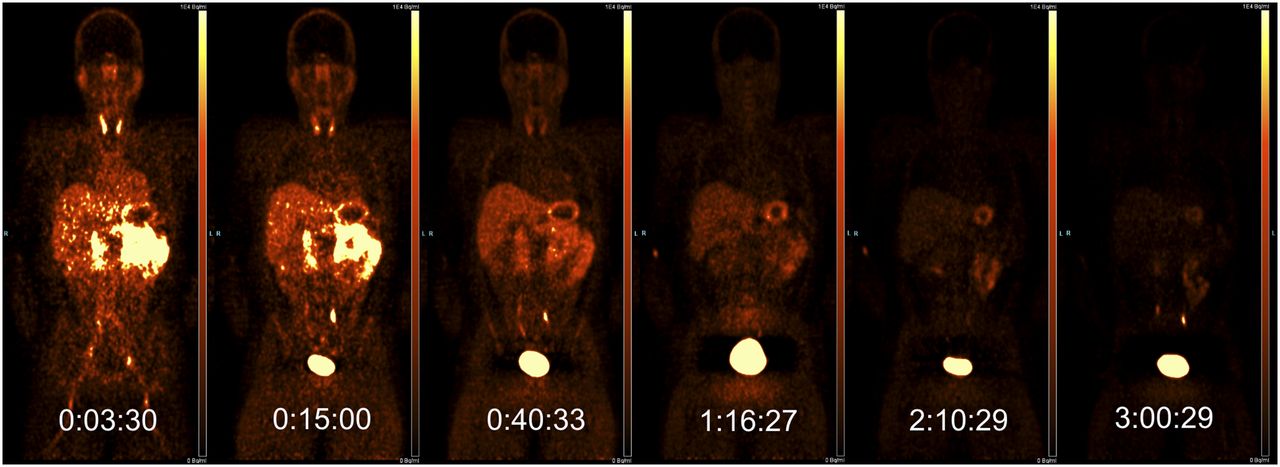
PET activity though single coronal slice for subject 1, over 6 scans (scale, 0–10 kBq/mL; mid-frame time points are shown in hours:minutes:seconds).
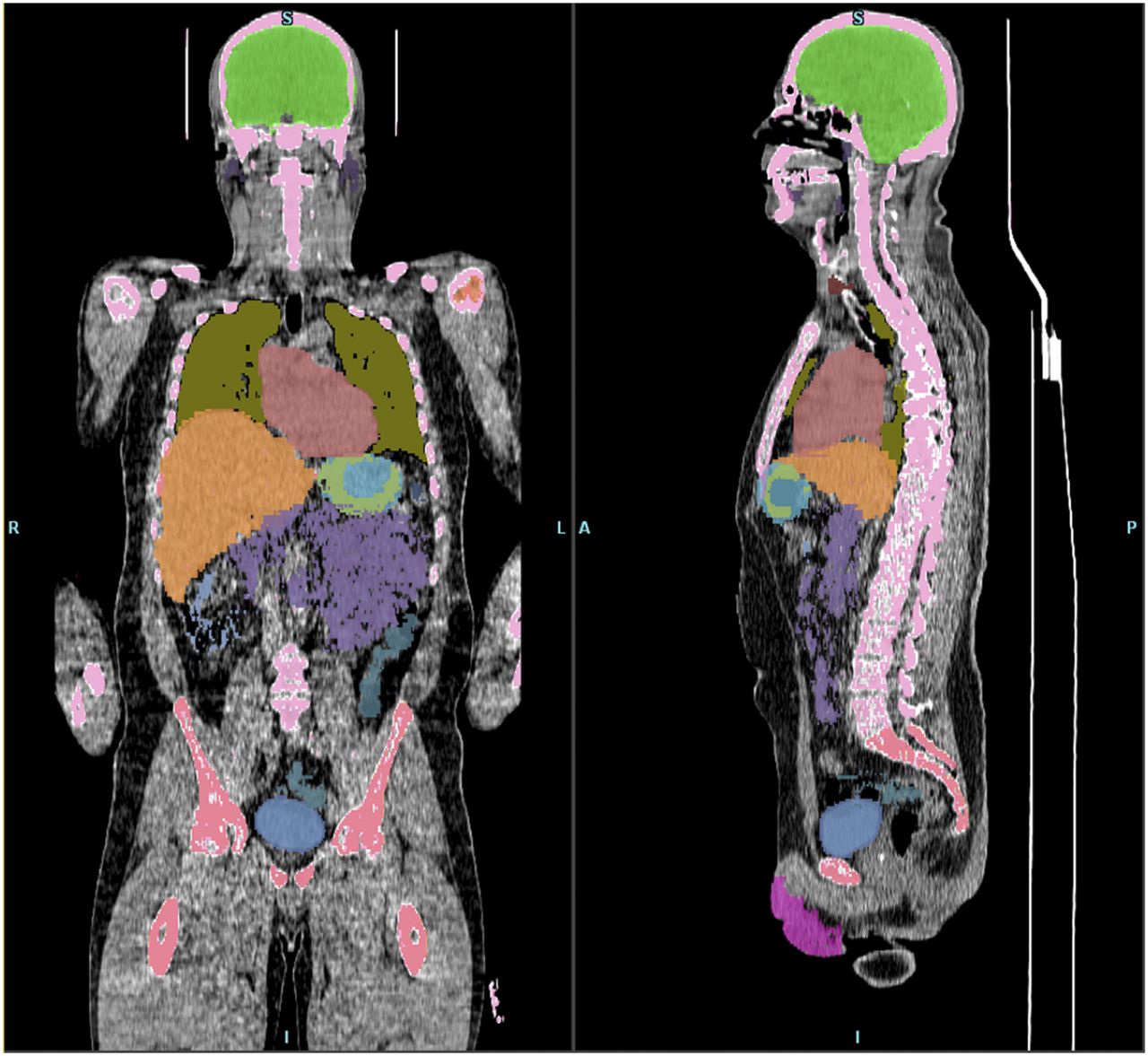
Organ ROIs for subject 1. Organs are colored to match those in time–activity curves (Fig. 3).
Time–activity curves from the ROIs through the 13 most active organs are shown in Figure 3 (colors of organ ROIs and time–activity curves are matched; time–activity curves with a maximum value of less than 2 kBq/mL are not shown and include muscle, testes, cortical bone, trabecular bone, bone marrow, lungs, and brain). The greatest activity concentration was observed for the bladder content, which, in contrast to the other organs, increased after the first scan, with the interruption of the void at 2 h. The other notable organs of uptake included the kidneys, small intestine, stomach wall, liver, spleen, thyroid, and gallbladder, whose time–activity curves generally had their highest point in scan 1 and decreased broadly linearly over this logarithmic graph, indicating exponential clearance. The time–activity curves for all 4 subjects were broadly similar.

Organ time–activity curves for subject 1.
Organ residence times for OLINDA source organs that could be identified from scan images for each subject, as well as the mean across all subjects, are shown in Table 2. Residence times for breast tissue were assessed for female subjects only, and testes for males only. Organ total is the sum of the residence times measured in individual organ ROIs. Voided urine is calculated from the quantity of urine voided according to the modeled bladder fit. The largest component of the mean residence time was the voided urine, followed by remainder, muscle, bladder, and liver. The residence time was affected not only by the activity concentration but also by the organ mass—hence, the influence of larger organs such as muscle and liver.
Residence Times of 18F-FB-A20FMDV2 for Each Organ
The mean residence times were entered into OLINDA/EXM, and the results are in Table 3, showing the organ β, γ, and total absorbed doses (mGy) and effective dose contribution (mSv) from each organ, as well as the total effective dose per injected megabecquerel. The β-dose to the stomach wall was adjusted to correct for OLINDA/EXM’s use of the stomach contents as a source organ, rather than the wall itself.
Absorbed and Effective Dose to Each Organ
The critical organ was the urinary bladder, with an absorbed dose of 0.198 mGy/MBq. The effective dose derived from the mean of the organ residence times was 0.0217 mSv/MBq. The effective dose derived using each subject’s residence times ranged from 0.0217 to 0.0247 mSv/MBq, resulting in an SD of 0.0020 mSv/MBq.
DISCUSSION
To the best of our knowledge, this study involved the first-in-human administration of 18F-FB-A20FMDV2, a PET ligand that has been shown preclinically to bind with high affinity and specificity to the integrin αvβ6 (M. Onega et al., unpublished data, 2017; (21,28–30)). αvβ6 is involved in the etiology and pathogenesis of cancer and fibrosis, and its expression levels have prognostic and theranostic implications for patient management and drug development. Thus, a PET ligand that can be used to noninvasively measure expression levels of this integrin in humans is likely to have broad clinical application both in patient management and in the development of new treatments that target αvβ6. The current study provided information about the safety, tolerability, and biodistribution of 18F-FB-A20FMDV2 in healthy subjects, as well as the radiation dosimetry of this ligand.
The observed benign profile in the 4 humans tested supports the lack of findings in the preclinical toxicology tests. In addition, no antibodies to the ligand were detected either before or after administration of the ligand.
Immunohistochemistry in various mouse organs, using a monoclonal antibody specific for αvβ6, revealed that this integrin is expressed constitutively at high or moderate levels in the epithelium of the gallbladder, stomach, duodenum, ileum, and colon (31). Low levels of αvβ6 expression were detected in the mouse skin (31). These data were supported by SPECT imaging data using indium-labeled A20FMDV2, but the lung was added to the list of organs expressing αvβ6 (31). PET images from the current human study showed focal uptake in the small intestine, stomach, liver, spleen, thyroid and gallbladder, but most of the activity was cleared to the bladder by the kidneys. The largest organ doses were within the abdominal cavity, with the highest value being to the bladder wall and then the kidneys, liver, small intestine, uterus, stomach, and spleen.
The effective dose, which was 0.0217 mSv/MBq, was lower than the figure from rat data (0.0335 mSv/MBq) (M. Onega et al., unpublished data, 2017), largely resulting from the lower residence time in the bladder and small intestine in humans, who have longer residence times than rats. The result is fairly typical for an 18F-based ligand and is close to the value of 0.019 mSv/MBq (32) for the most widely used PET ligand, 18F-FDG. The critical organ is the urinary bladder, which is common for a ligand that is cleared from the body by a renal route.
CONCLUSION
18F-FB-A20FMDV2 is a safe PET radioligand for future clinical studies to investigate changes in αvβ6 receptor availability as a result of increased expression in disease or after direct competition with a drug candidate. It has the potential to be used as a diagnostic, prognostic, and theranostic marker in the clinical management of cancer and fibrosis. In addition, 18F-FB-A20FMDV2 may be used in the development of novel therapies that target αvβ6.
DISCLOSURE
This study was sponsored by GlaxoSmithKline R&D (study numbers 200262 and NCT02612051). GlaxoSmithKline reviewed the manuscript for factual accuracy. Nicholas Keat, Mayca Onega, Will Hallett, Azeem Saleem, and Jan Passchier are employees of Imanova Ltd. Julia Kenny, Keguan Chen, Nadia Garman, Robert J. Slack, Christine A. Parker, Tom Lumbers, and Pauline T. Lukey are employees (or were employees at the time of the study) of GlaxoSmithKline R&D and own (or owned) shares in the company. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank the staff at Imanova Ltd. and GlaxoSmithKline R&D, Ware, Upper Merion, and Stevenage, for their professional conduct and successful delivery of all study procedures. We acknowledge the work of Andy Brown in the initial aspects of the study setup procedures.
Footnotes
Published online Feb. 2, 2018.
REFERENCES
- Received for publication October 10, 2017.
- Accepted for publication December 6, 2017.




{kind=link}
{kind=link}
{kind=link}