


Abstract
The aim of the present work was to validate a paper chromatography system as an alternative way to determine the radiochemical purity of Na18F. Methods: The evaluated parameters were specificity, limit of quantification, measurement interval, linearity, precision, accuracy, and robustness. Results: The proposed method proved to be linear (P > 0.05; r2 = 1.000), precise (relative SD, 8.6%), accurate (mean recovery, 95.9%; relative SD, 1.5%–1.8%), and robust under different conditions since no influence of the operative variables on the chromatographic performance was observed. Conclusion: This system can be used as a reliable alternative method to determine the radiochemical purity of Na18F samples that can be easily performed in PET radiopharmacies at low cost.
The use of Na18F for bone scintigraphy dates back to the early 1960s (1). However, the unavailability of clinical cyclotrons and the development of 99mTc-labeled agents for bone scintigraphy brought about the prompt replacement of Na18F with 99mTc agents for clinical use (2). Some decades later, Na18F was rediscovered when, in 2000, the Food and Drug Administration approved it as a radiopharmaceutical for bone scintigraphy as part of its modernization in the handling of new drug applications (3). Since then, many reports have proposed the use of this agent as a sensitive and specific radiopharmaceutical for detection of benign and malignant osseous abnormalities that also allows the regional characterization of lesions in metabolic bone diseases (4,5).
Na18F is a cyclotron-produced radiopharmaceutical, and the only reported and validated methodology for determining its radiochemical purity (RP) is high-performance liquid chromatography, which requires special equipment (6–8). Nevertheless, previous studies demonstrated the efficacy of a paper chromatography method for the RP determination of Na18F (9). This work was performed to investigate the possibility of applying the paper chromatography method as an alternative to high-performance liquid chromatography for 18F-fluoride RP analysis.
MATERIALS AND METHODS
Na18F Production
18F-fluoride was produced on an 18-MeV cyclotron, Cyclone 18/9 (IBA), by the 18O(p,n)18F nuclear reaction. The niobium target (with yield of 8.7 GBq/μA at saturation) was filled with 2 mL of enriched 18O water, which was irradiated with protons for 10 min at anintensity of 40 μA. The solution containing 18F− was transferred into an automatic synthesis module, Synthera (IBA), prepared with a commercial reagent kit and accessories for Na18F production. 18F-fluoride ions were trapped in an anion exchange column (Sep-Pak Light Accell Plus QMA; Waters Corp.) and were theneluted with a 0.9% NaCl solution. Finally, the resulting 15 mL of Na18F were dispensed into a sterile, pyrogen-free vial through a 0.22-μm filter (Millipore) in a dispensing unit (Dispensing Hot Cell; Becquerel and Sievert Co., Ltd.).
Physicochemical Quality Control
Radionuclidic purity was evaluated by γ-ray spectrometry (PX4 TeCd detector; Amptek).
Radionuclidic identity was determined by estimation of the half-life of 18F, which was calculated after measuring the radioactivity decay of the sample over a 20-min period in a radioisotope dose calibrator (Vexcal AV-02; Veccsa S.A.). The equation used is shown below:T1/2= ln2 (t − t0)/ln (a/a0), where T1/2 is half-life, t–t0 is the time interval (in minutes), a is activity measured after 20 min, and a0 is initial activity.
The pH of Na18F was measured using indicator strips of different ranges (universal indicator, pH 1–14, and special indicators, pH 2.5–4.5, 4.0–7.0, and 6.5–10.0; Merck) depending on the pH range of the samples. The results were compared with standard pH buffer, and the estimated value was registered.
Validation of Chromatographic Studies
The paper chromatographic system under evaluation was previously reported by Noto and Nicolini (9). Briefly, 5 μL of Na18F samples were spotted on a 15-cm strip of Whatman 1 paper as the stationary phase and developed with 0.15 M sodium acetate as the mobile phase for 30 min. Under these conditions, 18F− ion will be located at an Rf of 0.9–1.0, and possible impurities will be located at an Rf of 0.0–0.1. In this work, the methodology for Na18F RP determination was evaluated and validated by following international standards such as those of the International Committee of Harmonization (10) and the U.S. Pharmacopoeia (11). The evaluated parameters were specificity, limit of quantification, measurement interval, linearity, precision, accuracy, and robustness.
Linearity was determined using 18F in samples containing activity concentrations of 11.25% (sample A), 16.8% (sample B), 23.8% (sample C), 31.9% (sample D), 47.0% (sample E), and 100% (sample F). To test linearity, samples from the batch were diluted in 0.9% NaCl solution to reach the activity concentrations evaluated. One operator evaluated instrumental precision (as repeatability) by performing 6 replicates of 1 sample (sample E; 47% of activity concentration). Accuracy was assessed by performing 3 replicates for each of 3 samples having different activity concentrations (samples B, D, and F). Finally, robustness for chromatographic studies was evaluated by the variation of pH over 5 replicates of 2 samples having different final pH values: sample 1 (final pH, 4.0, with 1N HCl) and sample 2 (final pH, 8.0, with 1N NaOH), compared with control (pH, 5.8).
Each chromatogram was measured using an MS-1000F system (Eckert and Ziegler Radiopharma, Inc.), which consists of a Mini-Scan thin-layer radiochromatograph with a single-photomultiplier-tube Flow-Count integrator. The results for linearity, accuracy, and precision were obtained as total area under the main peak (counts). Linearity results included the equation obtained for the linear regression and its correlation coefficient (r2), with a P value of less than 0.05 considered statistically significant (12). Accuracy results are shown as the percentage of peak radioactivity recovered at an Rf of 0.9–1.0 for the different samples, including the percentage relative SD (RSD) for replicates. Instrumental precision results are shown as RSD, including the percentage coefficient of variation. Finally, robustness results are shown as variations in the Rf for replicates of samples 1, 2, and controls, with the results compared by ANOVA (13).
RESULTS
A total batch of 66.6 GBq of Na18F was produced in yield higher than 98%. The final activity concentration of the batch was 4.44 GBq/mL.
Physicochemical Quality Control
The γ-ray spectrum for radionuclidic purity assay showed only 1 main peak at 0.511 MeV. The radionuclidic identity of the final product, performed by the half-life estimation, was 110.2 ± 0.5 min, and its pH was 5.8.
Validation of Chromatographic Studies
The specificity of this chromatographic system has been previously demonstrated in a study that determined the Rf of pure fluoride and the ability of the system to separate the possible production impurities (9).
With regard to the other parameters, the regression curve proved linearity (r2 = 1.000; P > 0.05), with a y-intercept of 20,770 ± 13,060, a slope of 1,146,000 ± 268.5, and an absolute sum of squares of 1,537e+009 (Fig. 1). The accuracy studies demonstrated nearly 100% recovery (92.8%–98.2%; mean, 95.9%), with RSD values of 1.5%–1.8% for samples of the 3 activity concentrations (Table 1). In the repeatability studies (instrumental precision), the RSD was 8.6% (Table 2) and there were no significant differences in the recoveries obtained from replicates of sample E. The results of the robustness study showed that the operative variables had no influence on chromatographic performance, since differences in the pH of the Na18F product did not significantly affect the Rf of 18F− in the samples (Rf of 0.9–1.0 for all replicates of samples—both pH 4 and pH 8), with an RSD of 4.5% (data not shown).
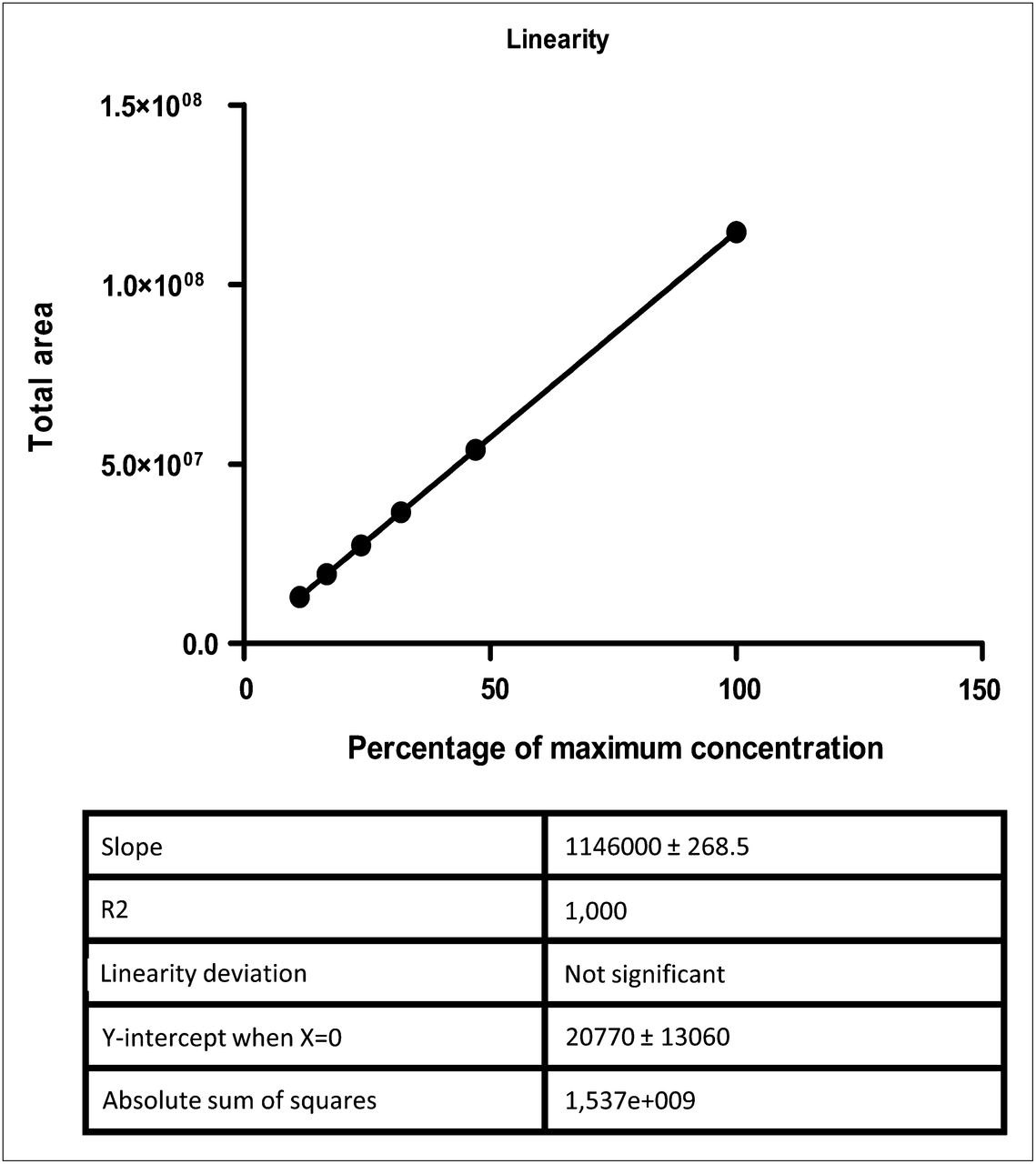
Linearity of chromatographic validation study.
Accuracy of Chromatographic Validation Study
Precision of Chromatographic Validation Study
DISCUSSION
Radionuclidic purity as evaluated by γ-ray spectrometry, and radionuclidic identity as determined by the half-life estimation and pH for Na18F, were in accordance with those found in the U.S. Pharmacopeia (7).
The limit of detection is the concentration derived from the smallest response that can be detected with reasonable certainty for a given analytic procedure (14). The detection threshold of a counting system is expressed in terms of background counting rates. The minimum detectable activity of a counting system is defined by the National Bureau of Standards as 3 SDs of the background counting rate, where the sample is counted for the same period. Thus, this value is associated with a 99.9% level of confidence that counts greater than the minimum detectable activity represent valid, detectable radioactivity. Nevertheless, in routine practice, sample volumes for measurements are selected in order to register at least 10,000 cpm (RSD, 1%) according to the efficiency of radioactivity detection of the equipment. Because radioactivity is a random phenomenon, activity measurements vary statistically and according to a Poisson distribution, and equipment often used for activity measurements can only estimate the real counts in a sample that is determined in a finite period. Therefore, it is not necessary to determine the limit of quantification—unlike high-performance liquid chromatography systems, for which injection volumes for samples are specified because of equipment requirements.
The measurement interval is defined as ranging from the background activity to the maximum measurable activity detected by the equipment under the proposed conditions of use. Background issues were discussed above in relation to the randomness of the radioactivity phenomena and the RSD often required in such measurements. Since the measurable maximum activity is defined and limited by factors such as saturation, but can be fitted in the linear range of the detector by changes in measurement efficiency (geometry), it does not need to be determined. Geometric efficiency is defined as the ratio of actually observed counts to the total number of γ-photons reaching the detector. In these terms, the efficiency of any scintillator detector eventually reaches a point at which it decreases as the activity of the sample measured increases (15,16). Nevertheless, coincidence losses may be avoided by changing the geometry of the sample with regard to the crystal or by placing a lead absorber between the detector and the chromatographic strip (15,16). This versatility in radiation measurements can be accomplished with this kind of equipment and RP methodology but is not possible in high-performance liquid chromatography methodology. Therefore, the interval will vary according to the equipment and measurement conditions selected for the chromatographic strip.
With regard to the other parameters validated, the regression curve proved linearity, accuracy, and precision according to the guidelines of the U.S. Pharmacopoeia and the International Conference of Harmonisation (10–11). The results of the robustness study demonstrated that neither ions such as Cl− and Na+ nor pH affected this parameter. Altogether, these results showed that the reproducibility of the measurements, characterized by the coefficient of variance of the recoveries, agrees with the expected performance parameters of thin-layer chromatography methods for assaying trace compounds (17).
CONCLUSION
The aim of the present work was to validate a paper chromatography system as an alternative way to determine the RP of Na18F. The proposed method proved to be linear, precise, accurate, and robust under different conditions and can easily be performed in PET radiopharmacies at low cost.
Acknowledgments
This work was financially supported by project UBACYT 20020100100489 from the University of Buenos Aires. Na18F was kindly donated by Laboratorios Bacon S.A.I.C., Buenos Aires, Argentina.
Footnotes
Published online Sep. 27, 2012.
REFERENCES
- Received for publication April 18, 2012.
- Accepted for publication June 26, 2012.




{kind=link}