


Abstract
A rapid quantitative kinetic chromogenic test in an automated portable test system has been developed for in-process and end-product determination of bacterial endotoxins in water using the Limulus amebocyte lysate. The aim of this work was to validate the method for 18F-FDG, 99mTc, and the lyophilized reagents methylene diphosphonic acid (MDP) and pyrophosphate for labeling with 99mTc radiopharmaceuticals with no interfering factors. Methods: Experiments were performed on 3 consecutive batches of 18F-FDG, 99mTc, MDP, and pyrophosphate produced at the Nuclear Energy and Research Institute of São Paulo, Brazil, using a portable test system. The maximum valid dilution (=500) was calculated to establish the extent of dilution to avoid interfering test conditions. Results: Better results were obtained above a 1:5 dilution factor for 18F-FDG and 99mTc, 1:20 for MDP, and 1:100 for pyrophosphate. The requirements of the test were satisfied (R ≤ 0.980, recovery of product positive control between 50% and 200%, and coefficient variation of samples < 25%), and the endotoxin concentration was lower than the lowest concentration of the standard curve (0.05 endotoxin unit mL−1) and therefore less than the established limit in pharmacopoeias. Conclusion: The portable test system is a rapid, simple, and accurate technique using the quantitative kinetic chromogenic method for bacterial endotoxin determination. For this reason, the test is practical for radiopharmaceutical uses and tends to be the method of choice for the pyrogen test. For 18F-FDG, 99mTc, MDP, and pyrophosphate, the validation was successfully performed.
Pyrogens include any substance capable of eliciting a febrile response on injection or infection. Endotoxin is a subset of pyrogens that are strictly of gram-negative origin, a natural complex of lipopolysaccharide occurring in the outer layer of the bilayered gram-negative bacterial cell. From the circulating blood cells of Limulus polyphemus, called amebocytes, a clear lysate is obtained that forms an opaque gel in the presence of extremely small concentrations of bacterial endotoxins (1).
Some techniques to determine endotoxins are known, such as the chromogenic (color development) and the turbidimetric (turbidity development) tests, both of which can provide valuable quantitative and qualitative information about the endotoxin concentration in samples (2).
The quantitative kinetic chromogenic method measures the chromophore released from a suitable chromogenic peptide by the reaction of endotoxins with the Limulus amebocyte lysate (LAL) reagent. The chromogenic method is based on bacterial endotoxin activation of the coagulating enzyme, which reacts with a colorless synthetic substrate. This substrate consists of a small peptide linked by the C-terminal arginine to the p-nitroaniline chromophore molecule. Once the cascading enzymatic reaction is activated, the coagulating enzyme causes the release of the yellow p-nitroaniline molecule. The color development is proportional to the endotoxin concentration in samples. The chromophore release can be spectrophotometrically monitored at 405 nm (optical density), and either the time to reach a predetermined absorbance of the reaction mixture or the rate of color development is measured (1–3).
A rapid quantitative kinetic chromogenic test in an automated portable test system has been developed for in-process and end-product determination of bacterial endotoxins in water using the LAL.
Preparatory tests should be conducted to verify that the criteria for the standard curve are valid, to confirm that the sample solution does not inhibit or enhance the reaction, and thus to ensure that the chromogenic technique is precise and valid for some specific sample (2,4). The aim of this work was to validate a rapid technique using the quantitative kinetic chromogenic method for determination of bacterial endotoxin in 18F-FDG, 99mTc, and the lyophilized reagents methylene diphosphonic acid (MDP) and pyrophosphate for labeling with 99mTc radiopharmaceuticals with no interfering factors.
MATERIALS AND METHODS
Experiments were performed on 3 consecutive batches of 18F-FDG, 99mTc, MDP, and pyrophosphate produced at the Nuclear Energy and Research Institute of São Paulo, Brazil, using a portable test system from Endosafe, Inc. Single polystyrene Endosafe cartridges (Figs. 1A and 1B) containing dry LAL reagents, control standard endotoxin, and synthetic color substrate were used.

(A) Endosafe cartridge scheme with 2 channels for sample negative control and 2 channels for sample positive control. (B) Sample channel zoom: 1 = sample negative control (sample + LAL + substrate) and 2 = sample positive control (sample + LAL + endotoxin + substrate).
The maximum valid dilution (MVD) is the maximum allowable dilution of a specimen at which the endotoxin limit can be determined. This dilution is applied to injections or to solutions for parenteral administration in the form constituted or diluted for administration. The general equation (5,6) to determine maximum valid dilution is as follows:
There are 3 defined concentrations in the archived kinetic chromogenic standard curve: 5.0, 0.5, and 0.05 EU mL−1 (2 logs). λ is the lowest point in the endotoxin standard curve—that is, the lowest endotoxin concentration that can be detected in the sample (2).
The maximum valid dilution obtained was 25 × 1.00, which, divided by 0.05, equals 500.
Serial dilutions of 1:1, 1:5, 1:10, 1:20, and 1:50 for 18F-FDG, 99mTc, MDP, and pyrophosphate, and also 1:100 and 1:200 for MDP and pyrophosphate, were prepared by dilution with sterile and pyrogen-free water. Samples (25 μL) were pipetted into the cartridge wells, and the temperature of the reaction was maintained at (37 ± 1)°C. The optical density was continuously monitored at 405 nm, and the overall time of the test was 15–20 min.
To be considered free of interfering factors under the conditions of the test, the measured concentration of the endotoxin added to the sample solution must be within 50%–200% of the known added endotoxin concentration after subtraction of any endotoxin detected in the solution without added endotoxin. The midpoint of the standard curve is the known added endotoxin concentration (0.5 EU mL−1) (2).
The test is not valid unless the following criteria are met: the product standard curve must meet the test for linearity, that is, correlation coefficient is equal to or greater than the absolute value of 0.980; the endotoxin recovery is within 50%–200%; the coefficient of variation is less than 25% on both sample and positive control channels; and the endotoxin concentration in the test product after correction for dilution and concentration is less than the endotoxin limit for the radiopharmaceutical (2).
Each test cartridge has 4 channels, 2 for the positive control, which contains a known endotoxin concentration, and 2 for the negative or sample control.
The following parameters were calculated by interpolation on an archived standard curve in the range of 5.0–0.05 EU mL−1, using the software in the sample reader: product endotoxin concentration (the amount of endotoxin in the product per volume after correction for dilution, expressed as EU mL−1), product positive control (the sample contaminated with a known endotoxin concentration [0.5 EU mL−1], expressed as EU mL−1), percentage sample coefficient of variation (the variation between the test samples in the 2 negative control channels), percentage endotoxin spike coefficient of variation (the variation between the test samples in the 2 positive control channels), and percentage recovery of product positive control (the measured concentration of the endotoxin added to the sample solution after subtraction of any endotoxin detected in the solution without added endotoxin).
RESULTS
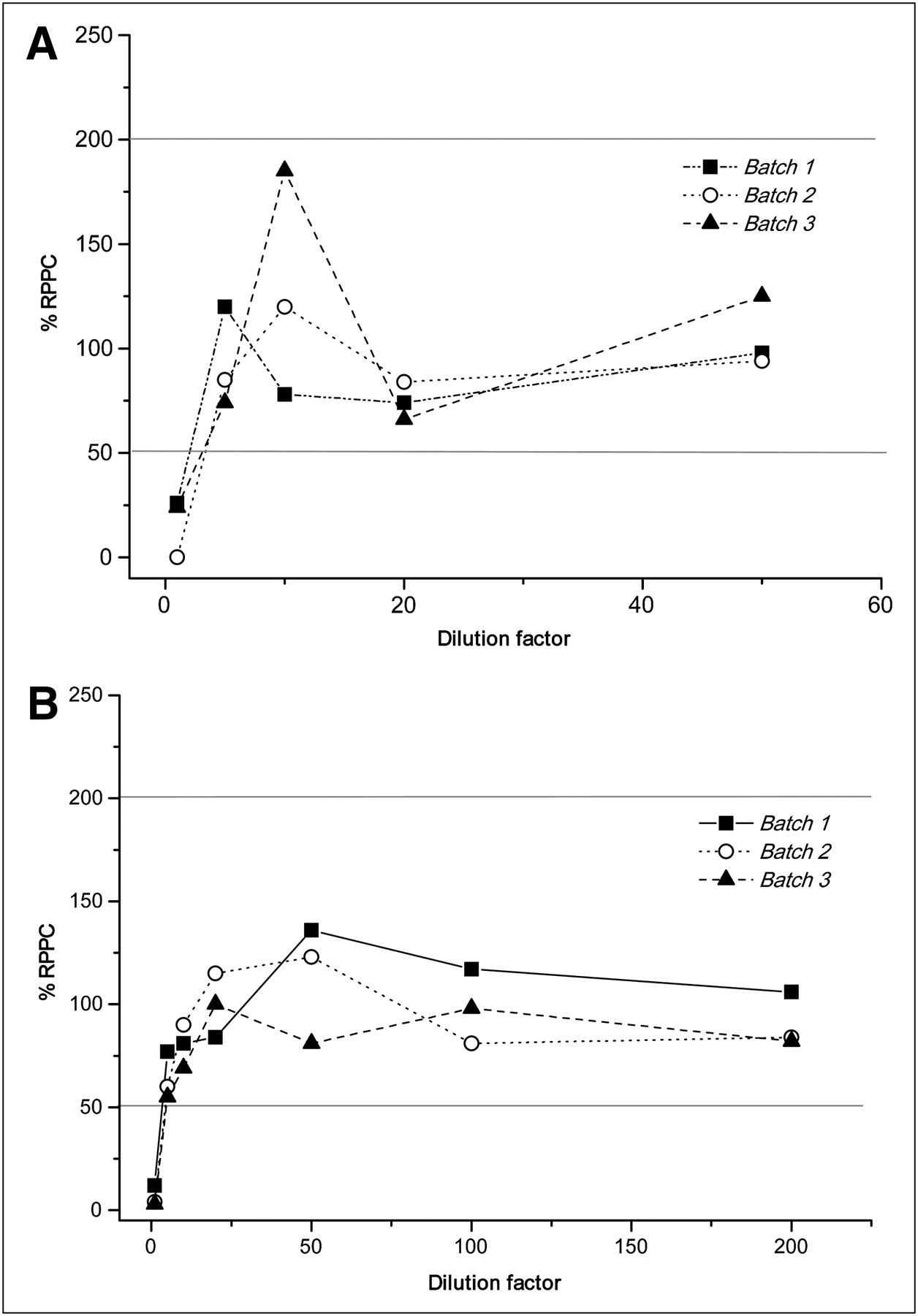
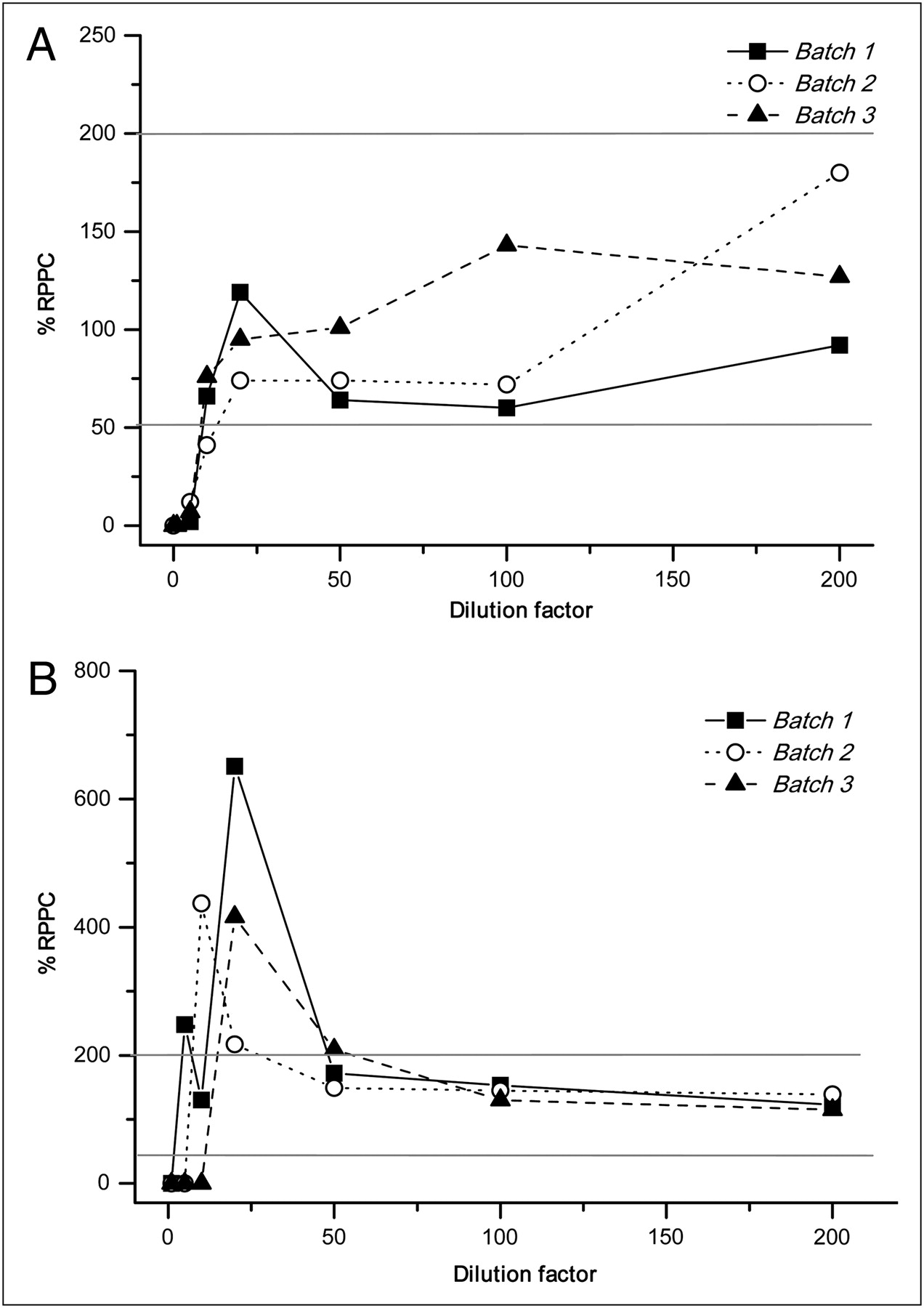
The percentage recovery of product positive control for serial dilutions of 3 batches of 18F-FDG and 99mTc, and of MDP and pyrophosphate, is shown in Figures 2A and 2B and Figures 3A and 3B, respectively.

Influence of dilution in percentage recovery of product positive control in 3 batches of 18F-FDG (A) and 99mTc (B) from undiluted to diluted samples. RPPC = recovery of product positive control.

Influence of dilution in percentage recovery of product positive control in 3 batches of MDP (A) and pyrophosphate (B) from undiluted to diluted samples. RPPC = recovery of product positive control.
Undiluted samples of each product interfered with the test. Experiments showed better results above a 1:5 dilution factor for 18F-FDG and 99mTc, 1:20 for MDP, and 1:100 for pyrophosphate.
Pyrophosphate showed significant interference below a 50× dilution; therefore, greater dilutions were performed.
DISCUSSION
To avoid interfering test conditions, the United States Pharmacopeia (6) allows drug product dilutions based on the established endotoxin limits, such as 175 EU per dose of radiopharmaceuticals. These limits may be used to determine the extent of dilution (maximum valid dilution) that may be applied to overcome an interference problem without exceeding the limit for endotoxin concentration (5–8).
In the chromogenic method, product dilution is higher than in the gel clot method (maximum valid dilution = 200 for radiopharmaceuticals with 0.125 EU mL−1 LAL reagent sensitivity), and the validation experiments were performed to determine the minimum interfering dilution.
Because of the linear quantitative correlation between log of the endotoxin concentration and log of the reaction time, product endotoxin concentration and product positive control can be determined when the assay parameters meet (9) the acceptability criteria presented in Table 1.
Acceptability Criteria for Kinetic Chromogenic Method (6)
An out-of-specification percentage recovery of product positive control is associated with a calculated product endotoxin concentration that expresses any interference (inhibition or enhancement) (9).When any criteria, mainly percentage recovery of product positive control, were not in the acceptable range, the test was not valid. Product endotoxin concentrations were obtained by multiplying the dilution factor by the lowest concentration of the standard curve (0.05 EU mL−1).
Because the sample volumes were small (25 μL), it was important to have skilled analysts to manage the micropipettes in order to minimize errors.
We observed that there was a specific dilution limit without interference for each radiopharmaceutical and that above that limit, the required parameters (R ≤ 0.980, recovery of product positive control between 50% and 200%, and coefficient of variation of samples <25%) were satisfied and therefore less than the established limit in pharmacopoeias. The percentage coefficient of variation of less than 5.0 showed good performance. The profile for the percentage recovery of product positive control versus dilution graph was similar in the 3 analyzed batches of each product.
CONCLUSION
The portable test system is a rapid, simple, and accurate technique using the quantitative kinetic chromogenic method for bacterial endotoxin determination. For this reason, the system is practical for radiopharmaceutical uses and tends to be the method of choice for the pyrogen test. For 18F-FDG, 99mTc, MDP, and pyrophosphate, the validation was successfully performed.
Acknowledgments
We thank the Nuclear Energy and Research Institute for providing test materials and quality control facilities.
- Received for publication July 22, 2010.
- Accepted for publication November 10, 2010.




{kind=link}
{kind=link}
{kind=link}