


Abstract
The understanding of key principles in pharmacology is essential to the clinical and research nuclear medicine practitioner. The scope of practice of the nuclear medicine technologist requires understanding of the indications, contraindications, warnings, precautions, proper use, drug interactions, and adverse reactions for interventional and adjunctive medications. This article is the companion to the third in a series of articles that aims to enhance the understanding of pharmacologic principles relevant to nuclear medicine. This article will build on the introductory concepts, terminology, and principles of pharmacology explored in previous articles in the series. Specifically, this article will focus on the pharmacologic principles associated with less commonly used interventional medications and adjunctive medications encountered in general nuclear medicine practice. Future articles will address the pharmacology related to nuclear cardiology, the emergency crash cart, and contrast media associated with CT and MRI.
A companion article to Part 3B, “Pharmacology, Part 3A,” appeared in the December issue of JNMT, page 326.
As outlined in the companion article (1), the scope of practice for nuclear medicine technologists (2) requires that they possess a thorough understanding and knowledge of indications, contraindications, warnings, precautions, proper use, drug interactions, and adverse reactions for each medication to be used. This scope demands sound understanding of the principles of pharmacology provided in earlier articles in this series (3,4). The content presented in these previous articles should be considered assumed knowledge, and those foundation principles will not be redefined here. The list of medications used either interventionally or adjunctively is long, and an exhaustive examination of all medications used in nuclear medicine is beyond the scope of this paper. Those common medications that are used adjunctively or are less common interventionally in general nuclear medicine will be outlined in detail. These have been summarized in Tables 1 and 2.
LESS COMMON INTERVENTIONAL STUDIES
There are several less frequently performed interventional procedures in general nuclear medicine. Acetazolamide evaluation of cerebral flow reserve and the use of H2 histamine antagonists or proton pump inhibitors to enhance detection of Meckel diverticulum are discussed below and summarized in Table 1. Glucagon and pentagastrin have been excluded, as these previously used approaches in Meckel diverticulum detection have been largely replaced by H2 histamine and proton pump inhibitors. Rarely used interventions for diabetic gastroparesis assessment have also been excluded (metoclopramide and erythromycin).
Acetazolamide (Diamox; Wyeth Holdings Corp.)
General Information/Drug Class
Acetazolamide is a sulfonamide carbonic anhydrase inhibitor (5–8). Regional cerebral blood flow and cerebral flow reserve can be assessed by perfusion imaging at rest and after vasodilation (8,9). Decreased flow reserve will not demonstrate the same increased flow in response to vasodilation as areas with a normal vascular supply (8,9).
Mode of Action
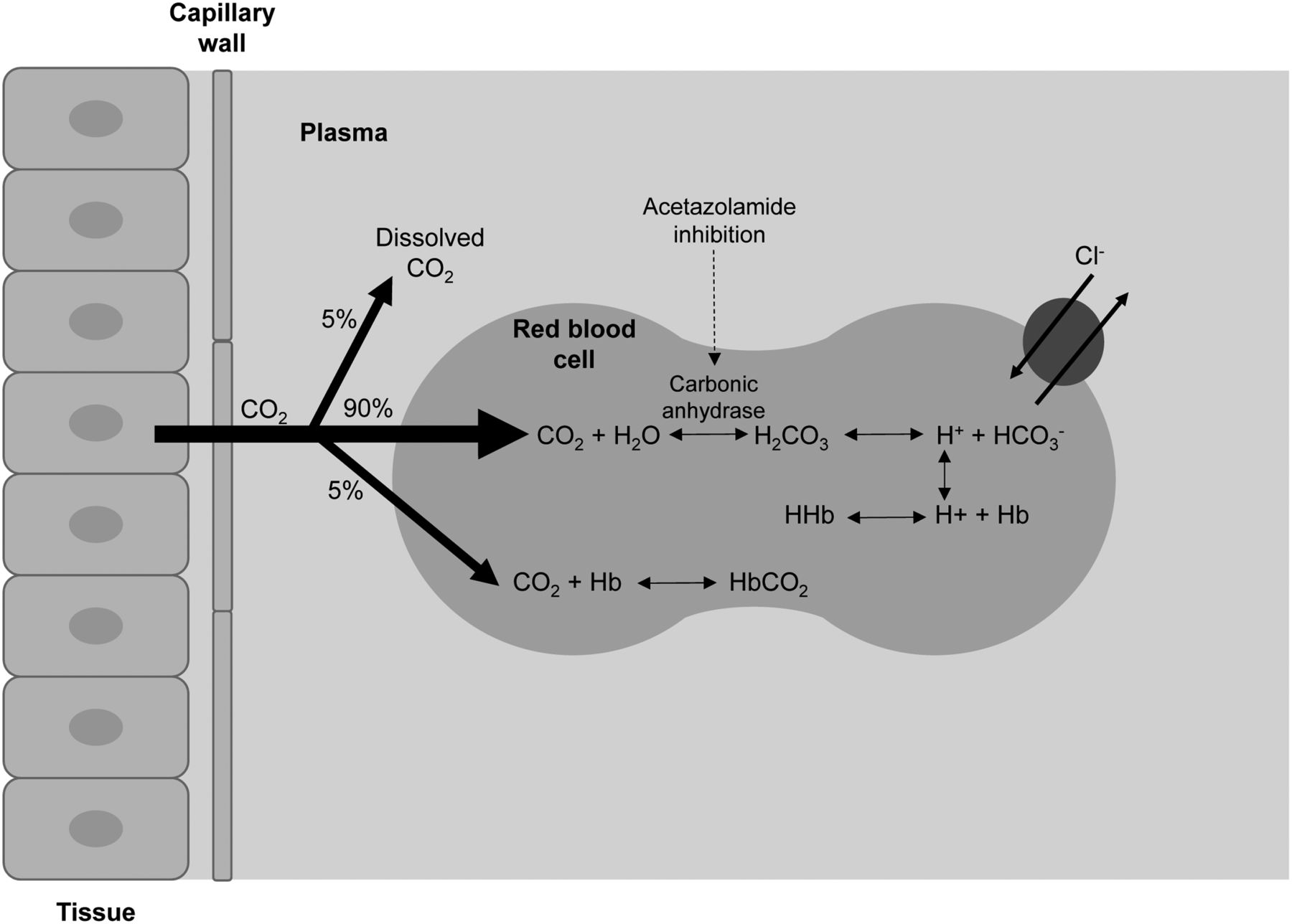
The predominant site for carbonic anhydrase is in the proximal tubule of the kidney (Fig. 1 in the companion article [1]), where it facilitates the carbon dioxide hydration/carbonic acid dehydration reaction (5,6,10,11). It is this reaction that controls bicarbonate reabsorption in the kidney. By inhibiting carbonic anhydrase, acetazolamide effectively causes sodium bicarbonate diuresis and an overall reduction of total-body stores of bicarbonate (6,10). Urinary alkalinization is linked to metabolic acidosis, and this results from the profound inhibition and blockade of bicarbonate reabsorption in the proximal tubule (5,6). Rapid and potent cerebral vasodilation follow acetazolamide injection because of the decrease in pH resulting from cerebral carbonic acidosis after inhibition of carbon dioxide clearance (12,13). The increased cerebral perfusion is due to intra- and extracellular acidosis (Fig. 1) (14). Cerebral flow response is not immediate, with a slow increase over a period of 15–25 min before returning to normal (15). This effect increases cerebral blood volume by 9% and cerebral blood flow by 50% in normal vascular territories (1,15,16). Although normal vessels readily dilate, diseased vessels do not, allowing the interventional study (compared with baseline) to exaggerate the blood flow difference between regions with normal and diseased vasculature (8,9,17).
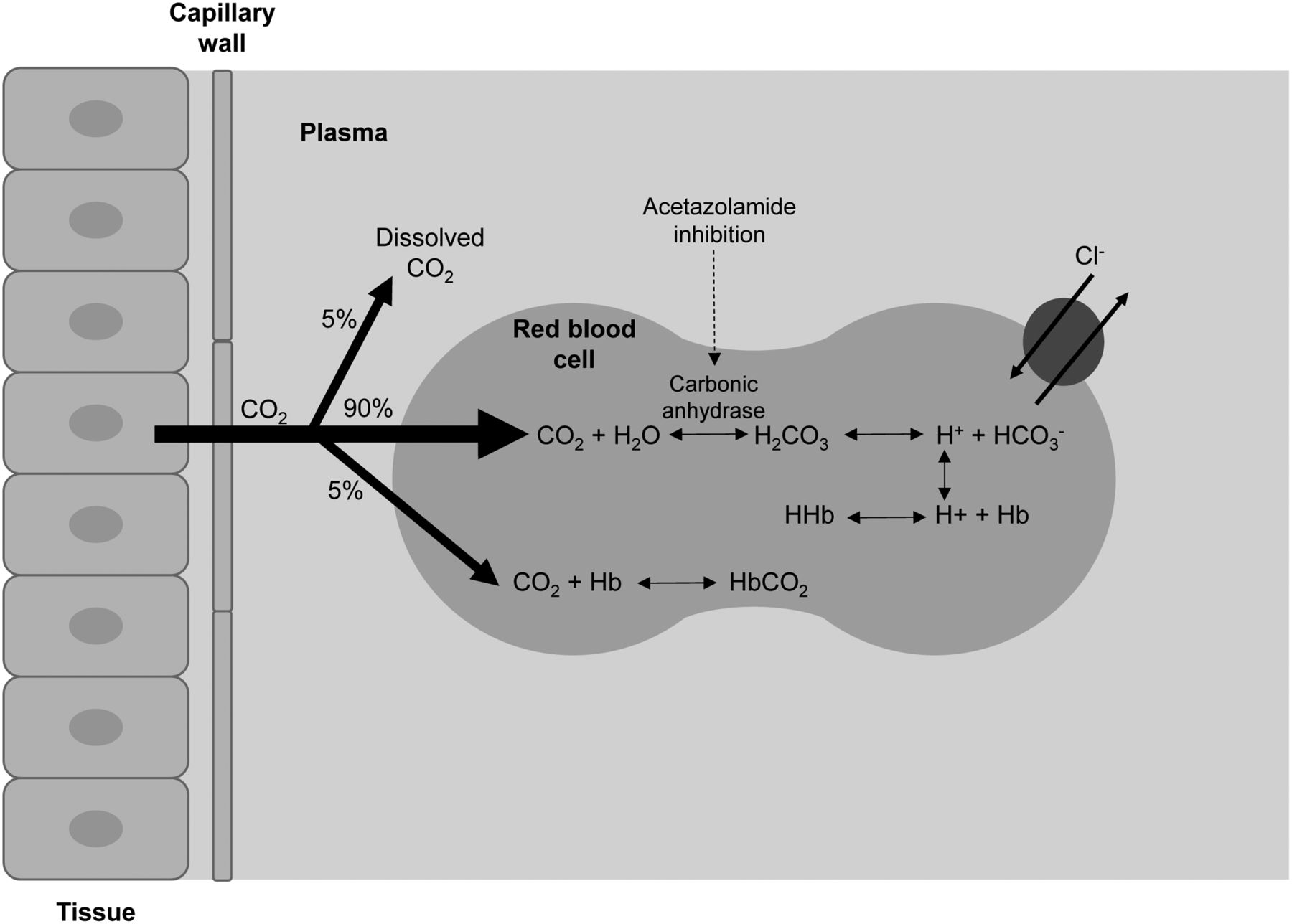
Schematic representation of impact of acetazolamide in inhibiting carbonic anhydrase and increasing serum carbon dioxide, which leads to increased cerebral perfusion.
Pharmacokinetics
Acetazolamide has an intravenous bioavailability of 100% (16,18). It has 70%–90% plasma protein binding, and 90% is excreted unchanged in urine (16,18). The elimination half-life is 3–6 h (8,9,16,18). After intravenous administration, onset of action occurs within 30 min, with peak activity seen by 2 h and a duration of effect of 12 h from a single dose (6,8,9,16–18).
Usual Indications
There are several other indications documented in the literature including use as a weak diuretic (6,11,16,17,19); in the acute management of glaucoma by minimizing the intraocular pressure (6,11,16,19,20); as second- or third-line therapy in epilepsy, particularly in absence seizures, tonic-clonic seizures, and myoclonic seizures (16,19); as a preferred agent for prophylaxis of altitude sickness syndrome (11,16,19); for the treatment of macular edema, Meniere’s disease, and neuromuscular disorders and to decrease intracranial pressure in idiopathic intracranial hypertension (16); and to decrease nephrotoxicity in patients taking methotrexate (16).
Use in Nuclear Medicine
Acetazolamide is used to increase cerebral blood flow for performing a stress test on the brain; for assessing cerebral flow reserve in patients with transient ischemic attacks, stroke, carotid artery stenosis, or other vascular disorders; and for differentiating vascular dementia from other causes of dementia (8,9,15,17).
Proper Use and Dose Administration
The standard dose is 1 g of acetazolamide diluted in 10 mL of sterile water, which is then administered by slow intravenous infusion (over 2 min) (8,9,17). Typically, this administration is done 25–30 min before the radiopharmaceutical administration (7,8).
Contraindications
Acetazolamide is contraindicated in cases of sodium or potassium depletion, hyperchloremic acidosis, Addison disease, adrenal insufficiency, and marked hepatic or renal impairment (16,19). It is also contraindicated in hepatic cirrhosis because of risk of hepatic encephalopathy (7,17). It should not be used in patients who have chronic closed-angle glaucoma (16).
Warnings and Precautions
Caution should be applied with acetazolamide use in known kidney or liver disease, diabetes, gout, lupus, hypotension, and pregnancy (16). Care should be taken when there is a known sulfonamide allergy (16,17). Acetazolamide can cause hyperglycemia in diabetic patients (16,17). Severe electrolyte imbalance can occur because of diuresis, severe renal disease may lead to nephrotoxicity, and caution should be exercised in patients with increased cerebral pressure (17). Acetazolamide is excreted in breast milk but need not be ceased even for therapeutic doses (as opposed to single interventional administrations) (16).
Adverse Reactions
For acetazolamide, any adverse reactions tend to be mild and transient but can include tingling sensations (extremities and mouth particularly), blurred vision, headache, dizziness, and confusion (5,7,8,17). Some patients experience flushing, nausea, tinnitus, and changes in taste (17). The diuretic effect can lead to urinary urgency (17). These adverse reactions can be minimized using the slow intravenous infusion (over 2 min). Longer-term use (therapeutic doses) can also lead to sun sensitivity, loss of appetite, increased body hair, hearing loss, tachycardia/arrhythmia, bleeding/bruising/hematuria, and mood changes (16).
Common Interactions
Acetazolamide has several important interactions that need consideration. First, it makes the urine alkaline and therefore decreases excretion of urine (16), potentially enhancing the effects of renally excreted or metabolized drugs (e.g., amphetamines and ciclosporin). Second, if used in conjunction with aspirin and other salicylates, acetazolamide can cause acidosis and central nervous system (CNS) toxicity (16). Third, if the patient is taking other antiepileptic drugs, the effects can be cumulative (16). Finally, alkalinization of the urine increases the solubility of methotrexate in the urine (16). Consequently, excretion of methotrexate is enhanced, and this effect can be used to decrease nephrotoxicity in those patients (16). There are several other reported interactions between acetazolamide and other medications potentially causing toxicity, including, but not limited to, carbamazepine, lithium, mexiletine, methadone, phenobarbital, phenytoin, and quinidine (21).
Cimetidine (Tagamet; GlaxoSmithKline)
General Information/Drug Class
The gastric mucosa concentrates 99mTc-pertechnetate, and this includes in Meckel diverticula (17). Cimetidine is an H2 histamine receptor antagonist that reduces gastric acidity (5,10,15,17,18,20). The detection of a Meckel diverticulum can be enhanced by either increasing the uptake at the site or by reducing migration of activity away from the site (15). Although the 99mTc-pertechnetate accumulation is not affected, the change in gastric acidity in response to cimetidine reduces the secretions into the gastric lumen, concentrating the radiopharmaceutical (7,17,20). This effect also applies to Meckel diverticula, enhancing visualization (17).
Mode of Action
Histamine is released from mast cells and stimulates H2 receptors in the gastric parietal cells to increase gastric acid secretion (5,6,10,18). Cimetidine is an H2 histamine antagonist that inhibits histamine stimulation of receptors (Fig. 2) and, thus, reduces both the volume and the concentration of hydrochloric acid in the stomach by as much as 50%–70% (5–7,10,17,18,20,22).
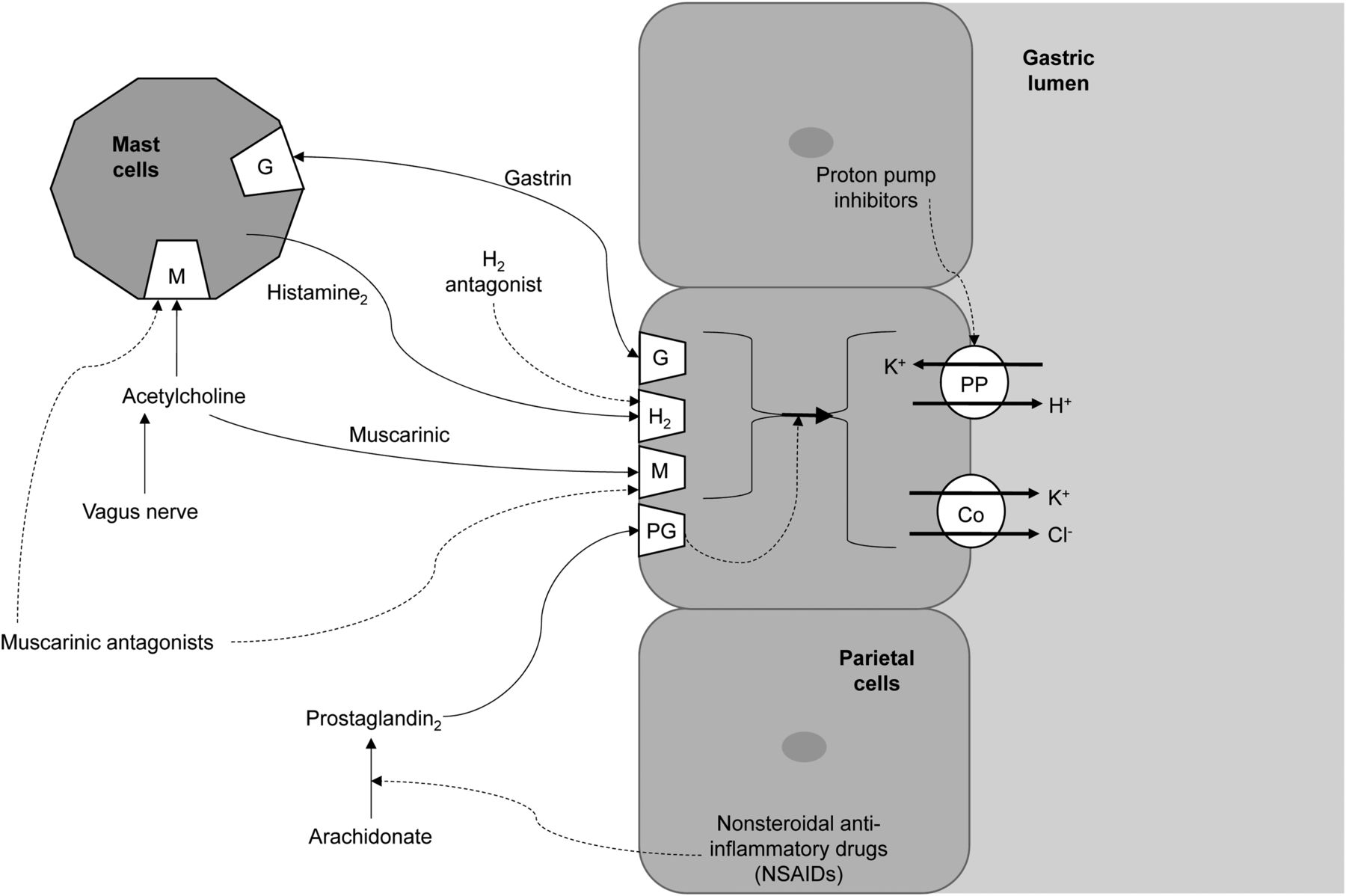
Schematic representation of action of receptors responsible for gastric acid secretion (solid lines) and inhibitors of gastric acid secretion (dashed lines). Co = cotransporter.
Pharmacokinetics
Cimetidine has an oral bioavailability of 60%–70% (16,18). It has 20% plasma protein binding and is partially (25%) metabolized in the liver, with 50% of the oral dose excreted unchanged in urine (16,18). The elimination half-life is 2 h (16,18), but this can be increased in renal dysfunction (16). After oral administration, onset of action occurs within 30–60 min, with peak activity seen at 60 min if taken on an empty stomach and 120 min with food, and significant effects last 4–6 h (16–18). Although cimetidine crosses the placenta and is excreted in breast milk, it is safe to use in both situations (16).
Usual Indications
Cimetidine is used to treat peptic ulcer disease, gastroesophageal reflux, dyspepsia, and stress ulcers by reducing gastric acid secretion (5,6,10,16,18). It has also been used for people at risk of aspiration during childbirth or general anesthesia (16).
Use in Nuclear Medicine
Cimetidine enhances radiopharmaceutical accumulation and visualization in Meckel diverticulum, improving the sensitivity of the test (17,20).
Proper Use and Dose Administration
Cimetidine can be administered orally as an 800- to 1,600-mg single dose 24 h before the Meckel diverticulum study (6,17). Alternatively, 300 mg orally, 4 times during the 24 h before the study, can be used (7,20). Oral administration is not always practical because of compliance issues associated with the regime, so a single intravenous dose of 300 mg in 100 mL of 5% dextrose for adults over 20 min (or 20 mg/kg in children) 1 h before the study can be used (7,17,20). The intravenous dose can be diluted in 20 mL of normal saline and administered over 2 min, but the adverse reaction rate might increase (17).
Contraindications
Known hypersensitivity to the medication is a contraindication for use (7).
Warnings and Precautions
Renal impairment should be managed by a reduced dose, and slow intravenous administration should be used in preference to rapid bolus administration for patients with cardiovascular impairment (16).
Adverse Reactions
Cimetidine is well tolerated and has a good safety profile (16,18). Nonetheless, patients may experience dizziness, confusion, headache, diarrhea, or bradycardia (5,10,16–18). Adverse reactions tend to self-resolve but can be limited by extending the intravenous administration over a longer period (e.g., up to 20 min) (17). Cimetidine has a weak antiandrogenic effect; gynecomastia, decreased libido, and impotence have been shown to occasionally occur in men, but these conditions are usually reversible (16).
Common Interactions
Cimetidine and other H2 antagonists can reduce the absorption of drugs reliant on gastric acidity, including dasatinib, ketoconazole, and itraconazole (16). Cimetidine decreases hepatic metabolism of warfarin, phenytoin, propranolol, nifedipine, chlordiazepoxide, diazepam, tricyclic antidepressants, lidocaine, theophylline, and metronidazole, thus increasing serum levels of these medications (7,16,18).
Ranitidine (Zantac; GlaxoSmithKline)
General Information/Drug Class
Ranitidine is an H2 histamine antagonist that reduces gastric acidity in the same way as cimetidine, discussed above. Ranitidine and cimetidine share the same drug class, general information, mode of action (Fig. 2), clinical use, use in nuclear medicine, contraindications, and precautions. Adverse reactions and common interactions have similarities but also some important differences that should be highlighted.
Pharmacokinetics
Ranitidine has an oral bioavailability of 50% (16,18). It has 15% plasma protein binding and is partially (4%–6%) metabolized in the liver, with 30% of the oral dose excreted unchanged in urine (16,18). The elimination half-life is 2–3 h (16,18), but this can be increased in renal dysfunction (16). After oral administration, onset of action occurs within 60 min, with peak activity seen at 2–3 h (independent of fasting) and significant effects lasting 4–13 h (16–18). Ranitidine crosses the placenta and is excreted in breast milk but is safe to use in both pregnancy and breastfeeding (16).
Proper Use and Dose Administration
Ranitidine is administered orally as a 300-mg dose to adults or as 1 mg/kg to a maximum dose of 50 mg intravenously over 20 min in children (6,20). The imaging procedure commences 1 h after ranitidine administration (20).
Adverse Reactions
The adverse reactions are the same for ranitidine as for cimetidine, except ranitidine does not have the androgenic effects associated with cimetidine (16).
Common Interactions
Unlike cimetidine, ranitidine does not affect cytochrome P450 and, therefore, has little effect on the metabolism of other medications in the liver (16,18). As with other H2 antagonists, ranitidine alters absorption of medications reliant on gastric acidity for absorption as outlined above for cimetidine (16).
Omeprazole
General Information/Drug Class
The detection of a Meckel diverticulum can be enhanced either by increasing the uptake at the site or by reducing migration of activity away from the site (15). Proton pump inhibitors decrease gastric acidity, increasing the availability of radiopharmaceutical for accumulation in the Meckel diverticulum (15).
Mode of Action
Omeprazole is a proton pump inhibitor that inhibits the secretion of gastric acid by inhibiting the proton pump (5,6,10,16,18). The proton pump is the enzyme system of hydrogen/potassium adenosine triphosphatase in the gastric parietal cell (Fig. 2) (5,6,10,16,18). In essence, when sufficient omeprazole accumulates or binds to the proton pump, the final step in acid production is inhibited, thus suppressing acid secretion (5,6,10,18). The action is irreversible inhibition, which is reflected in the long duration of effect relative to the elimination half-life (18).
Pharmacokinetics
Omeprazole is rapidly but variably absorbed after oral administration, with an oral bioavailability of 30%–40% (16,18). It has 95% plasma protein binding and is extensively metabolized in the liver, with 80% of metabolites excreted in urine and the remaining 20% by feces (16,18). The elimination half-life is 0.5 h (16,18), but this can be increased in renal dysfunction (16). After oral administration, onset of action occurs within 60 min, with peak activity seen at 120 min and significant effects lasting 3–5 d (16–18). Omeprazole has increased bioavailability in the elderly and those with liver dysfunction (16).
Usual Indications
Omeprazole is used for the treatment of peptic ulcer, nonsteroidal antiinflammatory drug–induced ulceration, esophageal erosion due to acid reflux, and Zollinger-Ellison syndrome (5,6,10,16,18). It is generally used in conditions for which gastric acid inhibition may relieve symptoms, including aspiration, dyspepsia, gastroesophageal reflux disease, and peptic ulcer (16). Proton pump inhibitors have also been used to reduce gastric acidity in the management of Helicobacter pylori (18).
Use in Nuclear Medicine
Omeprazole is used to enhance radiopharmaceutical accumulation in Meckel diverticulum by inhibiting the excretion from parietal cells (not affecting initial uptake).
Proper Use and Dose Administration
Omeprazole comes as a capsule formulation, which should be taken orally whole and not crushed, chewed, or opened (18). Therapeutically, omeprazole is administered as a single 20- to 40-mg dose daily for 4–8 wk (6,18). For Meckel diverticulum preparation and in consideration of pharmacokinetic information, omeprazole should be administered as a 40-mg dose on the morning preceding the scan and again on the day of the scan.
Contraindications
Omeprazole use is contraindicated in patients with known sensitivity (18).
Warnings and Precautions
The main precaution for omeprazole use is in patients with liver dysfunction (16,18). There is a potential risk of drug accumulation (due to a high degree of liver metabolism) (16,18); however, this risk is unlikely to present issues for isolated use as an interventional medication for Meckel diverticulum imaging.
Adverse Reactions
Omeprazole is well tolerated, and adverse reactions are minor (18). The risk of adverse reactions is further reduced with an isolated interventional dose. Nonetheless, the possibility of several adverse reactions should be considered, including abdominal pain, dizziness, headache, nausea, vomiting, diarrhea, flatulence, and rash (5,16,18).
Common Interactions
Omeprazole is metabolized by cytochrome P2C19 and cytochrome P3A4 and so can alter the metabolism of many other medications (16,18). Of note is the potential of omeprazole to increase plasma concentrations of diazepam, phenytoin, and warfarin (16,18). Omeprazole can also decrease absorption of medications reliant on stomach acidity, including atazanavir, dasatinib, ketoconazole, and itraconazole (16,18).
COMMON ADJUNCTIVES IN GENERAL NUCLEAR MEDICINE
There are several adjunctive medications used in general nuclear medicine, with the most common being sedatives, anxiolytics, laxatives, and anticoagulants. Specific medications used for each purpose will vary among clinical centers. Consequently, a prototype approach to medication selection will be adopted to allow a general understanding that can be translated for more specific application when alternative medications are used. For example, diazepam is the prototype anxiolytic medication and is used widely clinically and therapeutically; however, some sites may use alternative benzodiazepams (e.g., temazepam) or other drug classes (e.g., barbiturates). As outlined in Table 2, this discussion will focus on chloral hydrate as a sedative, diazepam as an anxiolytic, bisacodyl as a laxative, and heparin as an anticoagulant.
Chloral Hydrate
General Information/Drug Class
Chloral hydrate has a simple chemical structure that is similar to ethanol and chloroform (6,18). A combination of chloral hydrate with alcohol produces the potentially toxic additive effects known as “knockout drops” or a “Mickey Finn” (16,18).
Mode of Action
Chloral hydrate is a prodrug that is converted to trichloroethanol (5,18). It is a powerful hypnotic with CNS depressant properties that has been used for pediatric sedation in nuclear medicine, but its mode of action remains uncertain (5,18). It is thought to act by modulating the inhibitory properties of γ-aminobutyric acid (GABA) in neurotransmission (5).
Pharmacokinetics
Chloral hydrate is absorbed well from the gut and is rapidly metabolized to trichloroethanol in the liver (5). The elimination half-life is 7–11 h (16). After oral administration, onset of action (sedation) occurs within 30–60 min, with significant effects lasting 4–8 h (16).
Usual Indications
Chloral hydrate is used as a sedative, a hypnotic, and a premedication for medical procedures (5,6,16,18).
Use in Nuclear Medicine
In nuclear medicine, chloral hydrate is used for the sedation of pediatric patients for imaging procedures (18).
Proper Use and Dose Administration
The prescribed pediatric dose is 30–50 mg/kg to a maximum of 1 g as an oral liquid given 45–60 min before the procedure (16). Doses of 100 mg/kg up to 2 g can be used in children with respiratory monitoring (16).
Contraindications
Chloral hydrate is contraindicated in significant liver, kidney, or cardiac dysfunction (16).
Warnings and Precautions
Chloral hydrate should be used with caution in respiratory insufficiency (16). Hypersensitivity reactions can occur, as can paradoxic excitement. Because the effects can last more than 24 h, driving or using machinery should be avoided (16). Chloral hydrate is excreted in breast milk and can cause infant sedation (16).
Adverse Reactions
There are several common adverse reactions to chloral hydrate, including nausea, vomiting, diarrhea, dizziness, ataxia, drowsiness, lightheadedness, headache, hallucination, confusion, and paradoxic excitement (5,16). Hypersensitivity can lead to rash (16). Gastric irritation, abdominal distention, and flatulence have also been reported (16). It is possible with overdose to cause respiratory and cardiac depression (5,18).
Diazepam (Valium; Hoffmann-La Roche)
General Information/Drug Class
Benzodiazepams are widely used hypnotics and anxiolytics because of their superior safety profile, which includes reduced dose-related effects, fatality from toxicity and overdose, abuse potential, adverse effects, and serious drug interactions (5,6,18). Diazepam is the prototype diazepam, with newer variations having shorter durations of effect (e.g., lorazepam) and thus further improved safety profiles (6,18).
Mode of Action
Diazepam is a CNS depressant that has specific functions to reduce anxiety (5,6,10,18). Diazepam acts on the GABA receptor (5,6,10,18). GABA is an endogenous inhibitory neurotransmitter that causes increased chloride permeability, hyperpolarization, and decreased excitability of the neuron (5,6,10,18). Diazepam is not a GABA agonist but, rather, modulates the binding of GABA to GABA receptor A, further inhibiting the neuron (5,10,18).
Pharmacokinetics
Diazepam is a long-acting benzodiazepam, with metabolites that have their own activity (e.g., temazepam) (10,18). It has an oral bioavailability of almost 100%, being easily and completely absorbed after oral administration (16). It has 98%–99% plasma protein binding and is extensively metabolized in the liver, with excretion in urine in the form of conjugated or free metabolites (16,18). The elimination half-life is biphasic, with a rapid elimination phase followed by a second phase with a half-life of 1–2 d (16). After oral administration, onset of action occurs within 15–45 min depending on the presence or absence of food, with peak activity seen at 30–90 min and significant effects being prolonged by the 2–5 d half-life of active metabolites (16). Diazepam crosses the placenta and is excreted in breast milk; it should therefore be used with caution in pregnancy and breastfeeding (16).
Usual Indications
Diazepam is used for short-term management of anxiety or muscle spasm, as a premedication for sedation, and as an adjunctive medication for managing seizures in the acute setting (5,6,10,16,18).
Use in Nuclear Medicine
Diazepam is used to minimize anxiety in the claustrophobic patient, especially those undergoing SPECT, PET, and MRI.
Proper Use and Dose Administration
Diazepam can be administered intravenously, intramuscularly, rectally, and orally; however, for ease of use as an anxiolytic in nuclear medicine, the oral route is typically used. Doses should be individualized for age, liver and kidney function, and purpose, with typical doses ranging from 1 to 10 mg (16,18). If diazepam is being applied for sedation in adults, 10–20 mg intravenously over 2–4 min is used (16).
Contraindications
Diazepam is contraindicated in patients with chronic obstructive pulmonary disease, other severe respiratory diseases, severe liver disease, sleep apnea, glaucoma, myasthenia gravis, dependence on the drug, and hypersensitivity to the drug (16,18). Diazepam use should be avoided in those who already have CNS depression (16,18).
Warnings and Precautions
Diazepam should be used for only short periods, and withdrawal can be protracted. It should be used with caution in glaucoma, liver and kidney dysfunction, depression, psychosis, the elderly or very young, pregnancy, and breastfeeding (16,18).
Adverse Reactions
Adverse reactions have an increased risk of occurrence in the elderly (6). The most common adverse reactions to diazepam are drowsiness, sedation, muscle weakness, and ataxia (16). Less frequently, patients may experience vertigo, headache, confusion, and, with ongoing use, depression, dysarthria, changes in libido, tremor, visual disturbances, urinary issues, gastrointestinal disturbances, and amnesia (16). Paradoxic excitement is possible (16,18).
Common Interactions
Additive effects may be seen with other CNS depressants, including alcohol, sedating antihistamines, opioids, sedatives, and antidepressants (16,18). Many drugs metabolized in the liver, including H2 antagonists, can inhibit diazepam metabolism and thus prolong its effect (16,18).
Bisacodyl
General Information/Drug Class
There are several classifications for laxatives that might be used for interventional purposes (5,6,10,18). Fecal softeners (e.g., docusate) use wetting agents to mix water and fatty substances with the feces but have a 1- to 3-d onset of action. Bulk-forming agents (e.g., psyllium) cause water absorption, bowel distention, and reflex bowel activity, with a 12-h to 3-d lag to onset of action. Osmotics (e.g., lactulose) increase the volume of liquid in the bowel lumen, with onset in 1–3 h. Lubricants (e.g., liquid paraffin) coat the surface of the feces to aid passage, with a 6- to 8-h time to onset. Stimulants (e.g., bisacodyl) increase peristalsis via innervation, with an onset of 6–12 h; this category includes sodium picosulfate, typically used for bowel preparation for medical procedures. Combination therapy (e.g., softener and stimulant) can also be used.
Mode of Action
Bisacodyl is a stimulant laxative that increases water retention in the stool and stimulates peristalsis in the bowel (5,6,10,18). As such, it is effective for clearing the bowel of fecal content but should not be used regularly for constipation (5,6,10,18).
Pharmacokinetics
Bisacodyl has minimal absorption (16,18). The small amount that may be absorbed after oral administration has 99% plasma protein binding, is metabolized in the liver, and is eliminated via the kidneys (16,18). After oral administration, onset of action occurs within 6–8 h (16,18).
Usual Indications
The usual indications for bisacodyl are for preparation for medical procedures and for treatment of severe constipation (5,10).
Use in Nuclear Medicine
Bisacodyl is used to differentiate interluminal bowel activity from pathologic accumulation by stimulating fecal progression or passage in studies in which colonic activity can obscure pathology (e.g., 67Ga-citrate studies for non-Hodgkin lymphoma or abdominal infection, and 123I-metaiodobenzylguanidine abdominal imaging).
Proper Use and Dose Administration
The oral dose of enteric coated tablets should not be taken within 1 h of ingestion of milk or antacids (5,18). To avoid gastric irritation and abdominal cramping, the tablet should not be crushed or chewed (18). The oral dose is typically 5–15 mg as a single daily dose for constipation and up to 30 mg for bowel cleansing ahead of procedures.
Contraindications
Bisacodyl should not be used when the bowel is obstructed (10).
Warnings and Precautions
Because of liver metabolism of the small fraction absorbed, caution should be exercised in patients with liver impairment.
Adverse Reactions
There are few major adverse reactions other than the expected local reactions, which include gastric irritation and cramping (10,18). Fluid and electrolyte depletion is possible (16,18).
Common Interactions
Possible interactions with medications that change gastric acidity (e.g., H2 antagonists and proton pump inhibitors) may alter the effects of bisacodyl (16).
Heparin
General Information/Drug Class
There are 5 main categories of anticoagulant medications (5,6,10,18): vitamin K antagonists (e.g., warfarin), antithrombin III–dependent anticoagulants (e.g., fondaparinux), direct thrombin inhibitors (e.g., lepirudin), direct factor X inhibitors (e.g., rivaroxaban), and heparin and low-molecular-weight heparins.
Heparin is a substance comprising repeating units of a disaccharide attached to a central protein; as a result, this unfractionated form has a variety of molecular weights (5,6,10,18). Heparin is formed in mast cells of the lung, liver, and intestinal mucosa (5,6,10,18,19). Heparin used for human injection tends to be derived from either porcine mucosa or bovine lung (5,18). This is an important consideration for patients with cultural beliefs that may prohibit the use of products originating from these animals. Fractionated heparins are the lower-molecular-weight heparins separated for more predictable use (actions and adverse reactions).
Mode of Action
Heparin combines with antithrombin III, which is a naturally occurring clotting factor found in plasma (5,6,10,18,19). The heparin–antithrombin III complex has numerous actions (5,6,10,18,19). It inactivates thrombin factor IIa (most pronounced effect) and factors IXa, Xa, XIa, and XIIa. Inactivation of thrombin prevents fibrin formation and activation of factors V and VIII, and inactivation of factor Xa prevents conversion of prothrombin to thrombin, which prevents the formation of fibrin from fibrinogen.
Heparin is useful in preventing clot formation and growth and in vivo venous thrombosis, but it does not break down existing clots.
Pharmacokinetics
Heparin is inactive orally and has an intravenous bioavailability that is somewhat erratic (5,16). It is highly plasma protein–binding, and only a small fraction is excreted unchanged in urine (16,18). The elimination is dose-dependent, with a rapid saturation process at higher doses and slower renal elimination at lower doses (5,18). The elimination half-life, therefore, averages 1.5 h but ranges from 30 min for low doses to up to 6 h for higher doses (5,16,18). After intravenous administration, onset of action occurs rapidly, with significant effects lasting less than 3–6 h (5,16,18). Heparin does not cross the placenta (5,18).
Usual Indications
Heparin is used to prevent and treat venous thromboembolism (without actually breaking it down) and to prevent blood clots during surgery (prophylactically) (5,6,10,18,19). It is used intravenously in preference to oral warfarin because it has an immediate onset of action and can be reversed if necessary with protamine sulfate (18).
Use in Nuclear Medicine
Heparin is used in nuclear medicine to prevent blood clotting during blood labeling procedures or to maintain intravenous lines.
Proper Use and Dose Administration
The specific dose of heparin used will depend on the technique and local protocol. For example, some sites may use preheparinized blood tubes whereas others may add a small volume of heparin to a syringe containing blood. Caution should be exercised when considering the order of adding heparin because it can label to the radionuclide. A typical dose for blood labeling would be 10–15 units/mL of blood.
Contraindications
Heparin is contraindicated in patients with heparin hypersensitivity, acute bacterial endocarditis, and bleeding risk (e.g., recent surgery or childbirth, peptic ulcer, severe liver disease, severe hypertension, hemophilia, cerebral aneurysm, or hemorrhage) (5,6,18,19).
Warnings and Precautions
Heparin should be used with caution when patients are concurrently using dextran, dipyridamole, or thrombolytics (18). It should also be used with caution in asthmatic patients, patients with an allergy to animal proteins, and patients with liver dysfunction (18,19).
Adverse Reactions
Bleeding and hemorrhage are the most common adverse reactions to heparin (5,10,18,19). Hypersensitivity reactions can occur (5,10,19).
Common Interactions
Several important interactions can occur with heparin, including an increased risk of bleeding when used with other anticoagulants (e.g., aspirin and nonsteroidal antiinflammatory drugs), and this risk of hemorrhage is particularly concerning when heparin is used with thrombolytics, dextran, and dipyridamole (18).
CONCLUSION
Nuclear medicine technologists may encounter a wide variety of interventional or adjunctive medications—some in common use and others less so—in general nuclear medicine practice. An understanding of basic pharmacology for these drugs or class prototypes allows enhanced practice. Specifically, this deeper understanding of pharmacology, indications, contraindications, warnings, precautions, proper use, drug interactions, and adverse reactions for each medication helps to ensure that nuclear medicine technologists meet the minimum capabilities outlined in their scope of practice (2). This, in turn, translates to safer practice and better patient outcomes.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Aug. 23, 2018.
CE credit: For CE credit, you can access the test for this article, as well as additional JNMT CE tests, online at https://www.snmmilearningcenter.org. Complete the test online no later than March 2022. Your online test will be scored immediately. You may make 3 attempts to pass the test and must answer 80% of the questions correctly to receive 1.0 CEH (Continuing Education Hour) credit. SNMMI members will have their CEH credit added to their VOICE transcript automatically; nonmembers will be able to print out a CE certificate upon successfully completing the test. The online test is free to SNMMI members; nonmembers must pay $15.00 by credit card when logging onto the website to take the test.
REFERENCES
- Received for publication May 23, 2018.
- Accepted for publication August 2, 2018.




{kind=link}
{kind=link}