


Abstract
Pharmacology principles provide key understanding that underpins the clinical and research roles of nuclear medicine practitioners. Indeed, the scope of practice of the nuclear medicine technologist demands knowledge and understanding of indications, contraindications, warnings, precautions, proper use, drug interactions, and adverse reactions for each medication to be used. This article is the third in a series of articles that aims to enhance the understanding of pharmacologic principles relevant to nuclear medicine. This article will build on the introductory concepts, terminology, and principles of pharmacology explored in the first 2 articles in the series. Specifically, this article will focus on the pharmacologic principles associated with interventional medications encountered in renal and biliary imaging. A companion article (part 3B) will examine the less commonly used interventional medications in general nuclear medicine and prototype adjunctive medications. Future articles will address the pharmacology related to nuclear cardiology, the emergency crash cart, and contrast media associated with CT and MRI.
See the companion article to Part 3A, “Pharmacology, Part 3B,” in the March 2019 issue of JNMT.
Patients presenting to the nuclear medicine department may be taking medication that can interfere with the nuclear medicine procedure. Typically, these represent cessation medications, and the period of cessation is largely dependent on the half-life of the medication combined with a clinical decision about the patients’ health when that medication is withheld (e.g., other angiotensin-converting-enzyme [ACE] inhibitors when performing renal scanning in renovascular hypertension). This interference can, however, allow the nuclear medicine procedure to be enhanced by controlling the introduction of the medication, deliberately altering physiology, and examining that response (e.g., the role of morphine in contracting the sphincter of Oddi in biliary scanning). These represent interventional medications, which may also be referred to as imaging medications. Interventional medications not only enhance the physiologic process being evaluated but also can increase the sensitivity and specificity of the procedure (1,2). It may also be necessary to administer a medication to patients to alter their condition in some way but without deliberately influencing the physiologic response being examined (e.g., sedation of a patient to gain compliance). These are referred to as adjunctive medications and will be addressed in the companion article in an upcoming issue of the Journal of Nuclear Medicine Technology (3).
The scope of practice for nuclear medicine technologists (4) requires that they display a thorough understanding and knowledge of indications, contraindications, warnings, precautions, proper use, drug interactions, and adverse reactions for each medication to be used. That understanding and knowledge need a command of the principles of pharmacology provided in earlier articles in this series (5,6). Indeed, the content conveyed in these previous articles should be considered assumed knowledge for this article and those that follow in the series. To that end, those foundation principles will not be redefined here. The list of medications used as either interventional or adjunctive medications is long, and an exhaustive examination of all medications used in nuclear medicine is beyond the scope of this article.
The scope of practice for nuclear medicine technologists (4) defines interventional (imaging) medications as those medications used to evoke a specific physiologic or biochemical response in conjunction with diagnostic imaging or therapeutic procedures (e.g., captopril and dipyridamole). The same document (4) defines adjunctive medications as those medications used to respond to a patient’s condition during a nuclear medicine procedure (e.g., chloral hydrate and aminophylline). For interventional medications routinely used in renal and biliary imaging, this article will identify indications for their use, outline the dosage and administration modes, explain the mode of action and pharmacology, and describe precautions, contraindications, and adverse reactions. These characteristics have been summarized in Tables 1 and 2.
INTERVENTIONAL RENAL SCANS
Interventional renal scans are among the most commonly performed procedures in general nuclear medicine. Diuretic and ACE inhibition renal imaging (renogram) are the principle interventional applications discussed here and summarized in Table 1.
Furosemide (Lasix; U.S. Pharmaceuticals Holdings II, LLC)
General Information/Drug Class
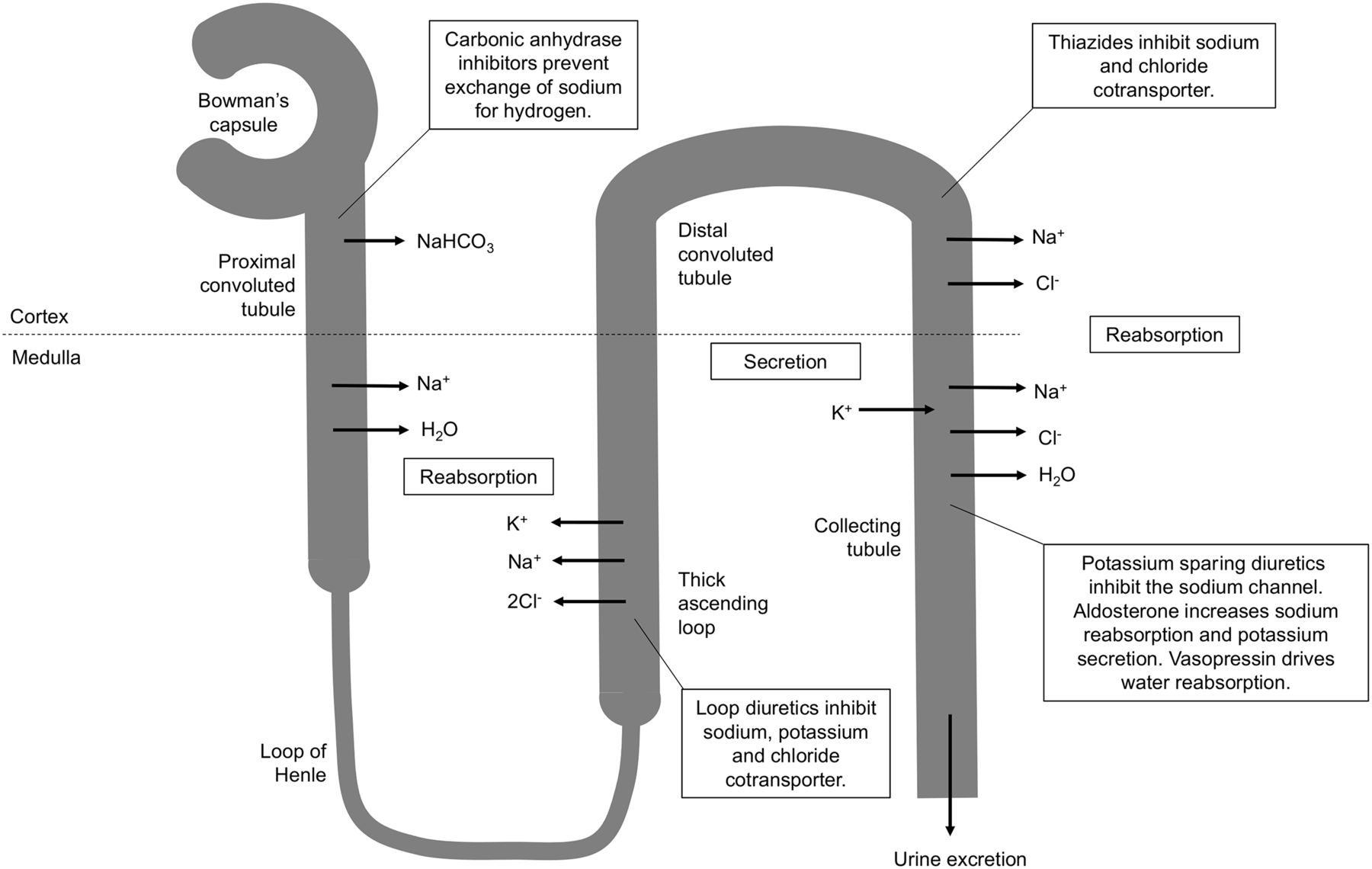
Diuretics generally increase urine volume by inhibiting reabsorption of sodium and chloride in the nephron (7–11). There are 3 main classes of diuretics—loop diuretics (e.g., Lasix/furosemide), thiazide diuretics (e.g., hydrochlorothiazide), and potassium-sparing diuretics (e.g., amiloride)—and a fourth class, carbonic anhydrase inhibitors (e.g., acetazolamide), is added to this discussion because it has an interventional application in brain scanning (Fig. 1).
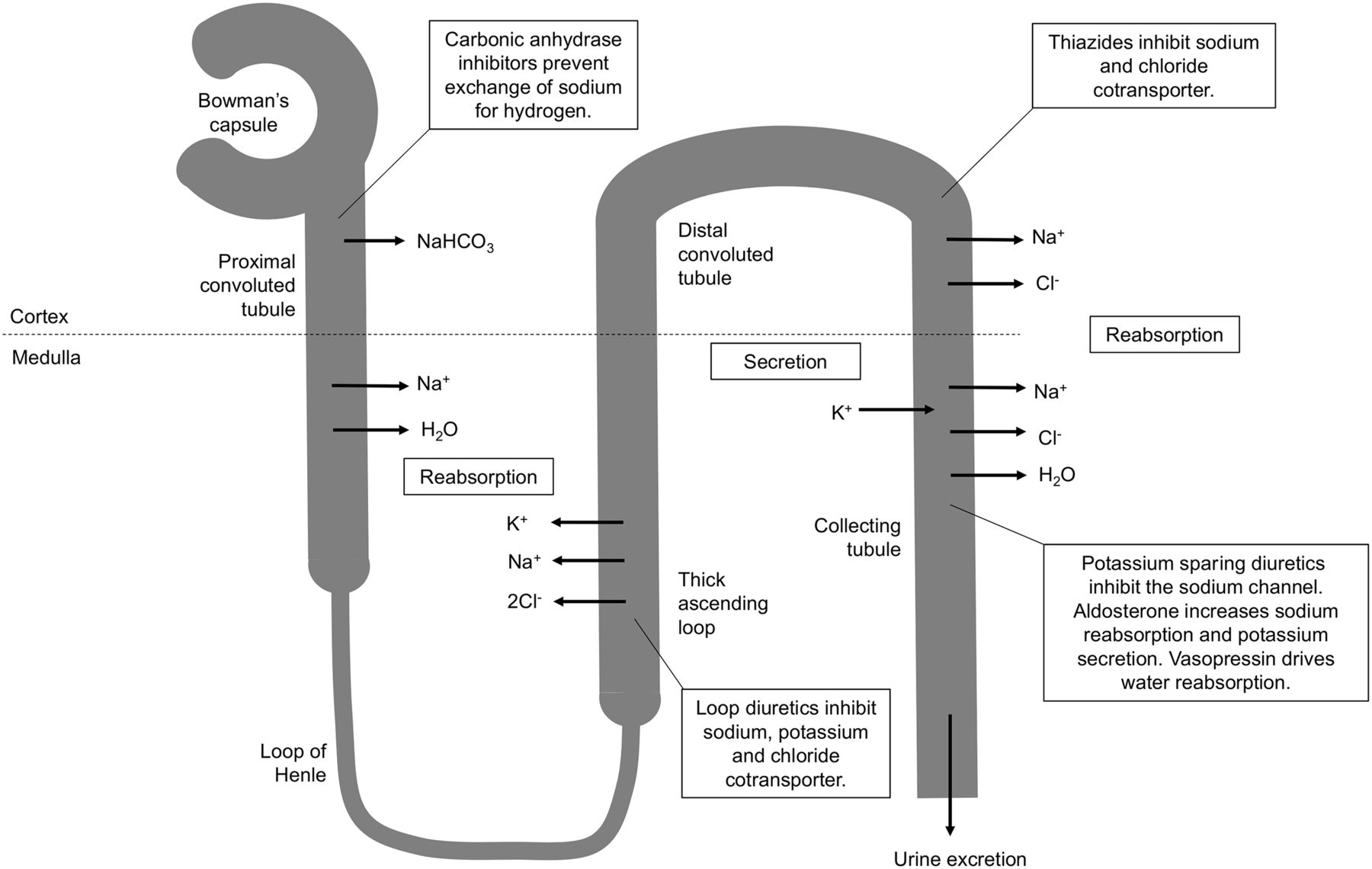
Schematic representation of sites of action of different classes of diuretics. Furosemide is loop diuretic acting on thick ascending portion of loop of Henle in nephron.
Furosemide is a sulfamoylbenzoic acid–based loop diuretic that increases urine volume by inhibiting reabsorption of sodium, potassium, and chloride (1,8–10,12). Although this can occur in proximal and distal tubules in the nephron, it is predominantly situated at the ascending aspect of the loop of Henle; hence the name loop diuretic (1,8–12). Loop diuretics are characterized by rapid onset and potent diuretic effects but typically of a short direction (7–9). Furosemide is active inside the lumen of the loop of Henle and, therefore, relies on adequate glomerular filtration and secretion of furosemide to have its effect (8–10). Consequently, furosemide may have a truncated response in renal impairment.
Increasing the flow rate of urine with furosemide allows differentiation of the obstructed kidney (postfurosemide clearance half-life greater than 20 min) from the unobstructed kidney (postfurosemide clearance half-life less than 10 min) and the dilated but not obstructed kidney (postfurosemide clearance half-life between 10 and 20 min) (13–15). Furosemide is the diuretic of choice because it has superior peak effects (15).
Mode of Action
There are 2 sodium (Na+), potassium (K+), and chloride (2Cl-) cotransporters (NKCC): NKCC1 and NKCC2 (7–10). The former is primarily involved in secretion and the latter primarily absorption; loop diuretics have an affinity for NKCC2 (7–9,11). Transport effectively relies on simultaneous binding to all 3 ion sites, and thus, blockade of just one results in inhibition (9,10). Furosemide binds to the chloride binding site and by doing so inhibits transport of sodium, potassium, and chloride in the loop of Henle (Fig. 1) (7–11,16). The increased urinary excretion of sodium, potassium, chloride, calcium, and magnesium results in low osmolality, which inhibits the reabsorption of water by the kidney, increasing urine volume (8–11,16). A secondary effect changes the charge difference across the lumen wall, inhibiting calcium and magnesium transport (8,9). Furosemide also releases vasodilating prostaglandins, which produce short-duration venodilation (8–11).
Pharmacokinetics
Furosemide has variable oral bioavailability (10%–100%), whereas intravenous administration provides 100% bioavailability (9). It has 95%–99% plasma protein binding, 50% is excreted unchanged in urine, and the remaining 50% is metabolized in the kidney (conjugated with glucuronic acid in the kidney) (8,9,16). The elimination half-life is 1.5–2 h (9), but this can be substantially extended in end-stage renal disease (10 h) and hepatic dysfunction (50–327 min) (16). After intravenous administration, onset of action occurs within 5 min, with peak activity seen at 15–30 min and significant effects lasting 2–3 h (1,8,9,16).
Usual Indications
The clinical role of furosemide tends to be to manage edema in congestive heart failure patients (1,2,7,8,10,11). It can also be used to treat hypertension (7,8,10,11,16).
Use in Nuclear Medicine
In nuclear medicine, furosemide is used to evaluate obstructive uropathy with renography (1,2,12).
Proper Use and Dose Administration
The standard adult intravenous dose is 20–40 mg, and pediatric doses are 0.5–1 mg/kg administered intravenously over 1–2 min (1,2,12,15). Given the time to peak activity, furosemide can be administered preemptively 15 min before the administration of the radiopharmaceutical. More typically, furosemide is administered 15–20 min after the radiopharmaceutical if radiopharmaceutical retention in the renal pelvis is apparent (1,2).
Contraindications
Furosemide is contraindicated in anuric patients, patients with known hypersensitivity (furosemide specifically or sulfonamides generally), and those significantly dehydrated or with sodium depletion (1,9,12).
Warnings and Precautions
Furosemide should be administered with caution to prevent injury or suture tearing in patients who have had recent urologic procedures (1). Patients should be adequately hydrated before receiving the drug (1,12). It should be used with caution in known kidney or liver disease, pregnancy, diabetes, gout, and lupus (16). Furosemide is excreted in breast milk (16).
Adverse Reactions
Adverse reactions to a single dose of furosemide are usually mild and transient and include nausea, vomiting, diarrhea, dizziness, hypotension, headache, tinnitus, rash, electrolyte imbalance, and dehydration (1,9,10). These adverse reactions are more common when furosemide is being used therapeutically. Therapeutic doses of furosemide may also cause vision disturbance, sun sensitivity, hearing impairment, confusion, arrhythmia, limb numbness, tingling and pain, yellow eyes and skin, and abdominal pain (7,16). Allergic reactions are possible (16). Adverse effects on hearing can be minimized with slower injection rates at 4 mg/min (16).
Common Interactions
Furosemide can enhance the nephrotoxicity of cephalosporin antibacterials and hearing impairment associated with aminoglycoside antibacterials (16). Furosemide can reduce the effect of antiepileptics such as phenytoin (16). A cumulative effect with other diuretics can lead to electrolyte imbalance and dehydration (16). Hypotension can be more severe when furosemide is used concurrently with ACE inhibitors or angiotensin receptor blockers (ARBs) (9). Furosemide can increase the risk of toxicity of lithium, digoxin, cisplatin, and aminoglycosides (9,16). Nonsteroidal antiinflammatory drugs (NSAIDs) decrease diuresis and can exacerbate side effects (9,16).
Captopril (Capoten; Bristol-Meyers Squibb)
General Information/Drug Class
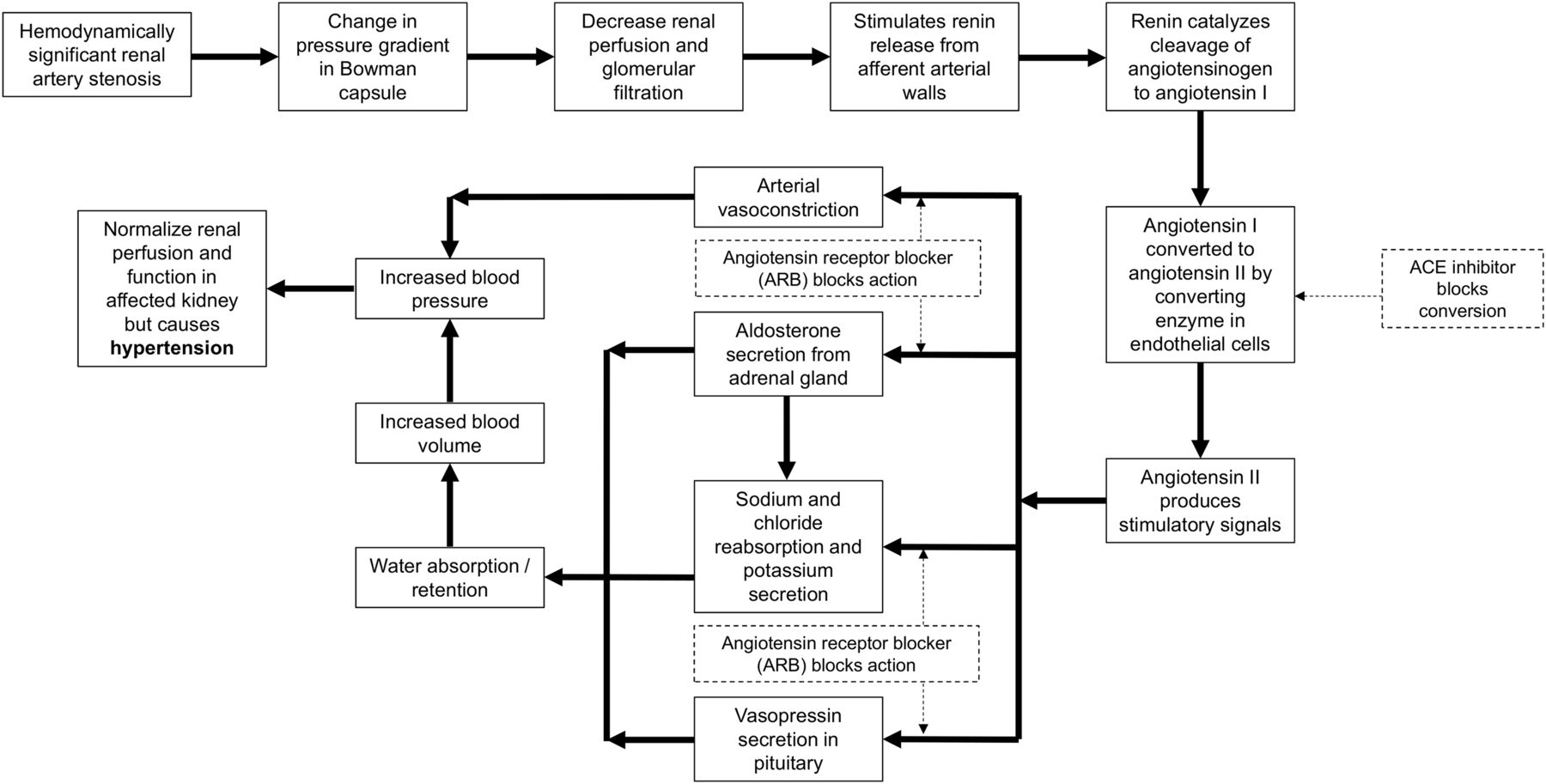
The kidneys play an important role in blood pressure regulation through the negative feedback mechanism of the renin-angiotensin-aldosterone system (7–11). A decrease in blood flow through the kidneys (decreased arterial blood pressure) triggers a pressure gradient change between afferent and efferent arterioles in the Bowman capsule (8–11), leading to reduced glomerular filtration and release of renin, which facilitates the production of angiotensin I (8–11). As illustrated in Figure 2, an ACE converts angiotensin I to angiotensin II, leading to vasoconstriction and increased blood volume, which, in turn, increases blood pressure (7–11). The presence of a hemodynamically significant renal artery stenosis initiates this cascade, resulting in restoration of glomerular filtration (near-normal renogram) and hypertension (8,9). A 75% stenosis is typically considered necessary for hemodynamic significance, although there is variability in reported cut-offs down to 50% stenosis. Bilateral renal artery stenosis has increased renin production from both kidneys, whereas unilateral disease has increased renin in the diseased kidney and suppressed renin production in the normal kidney.
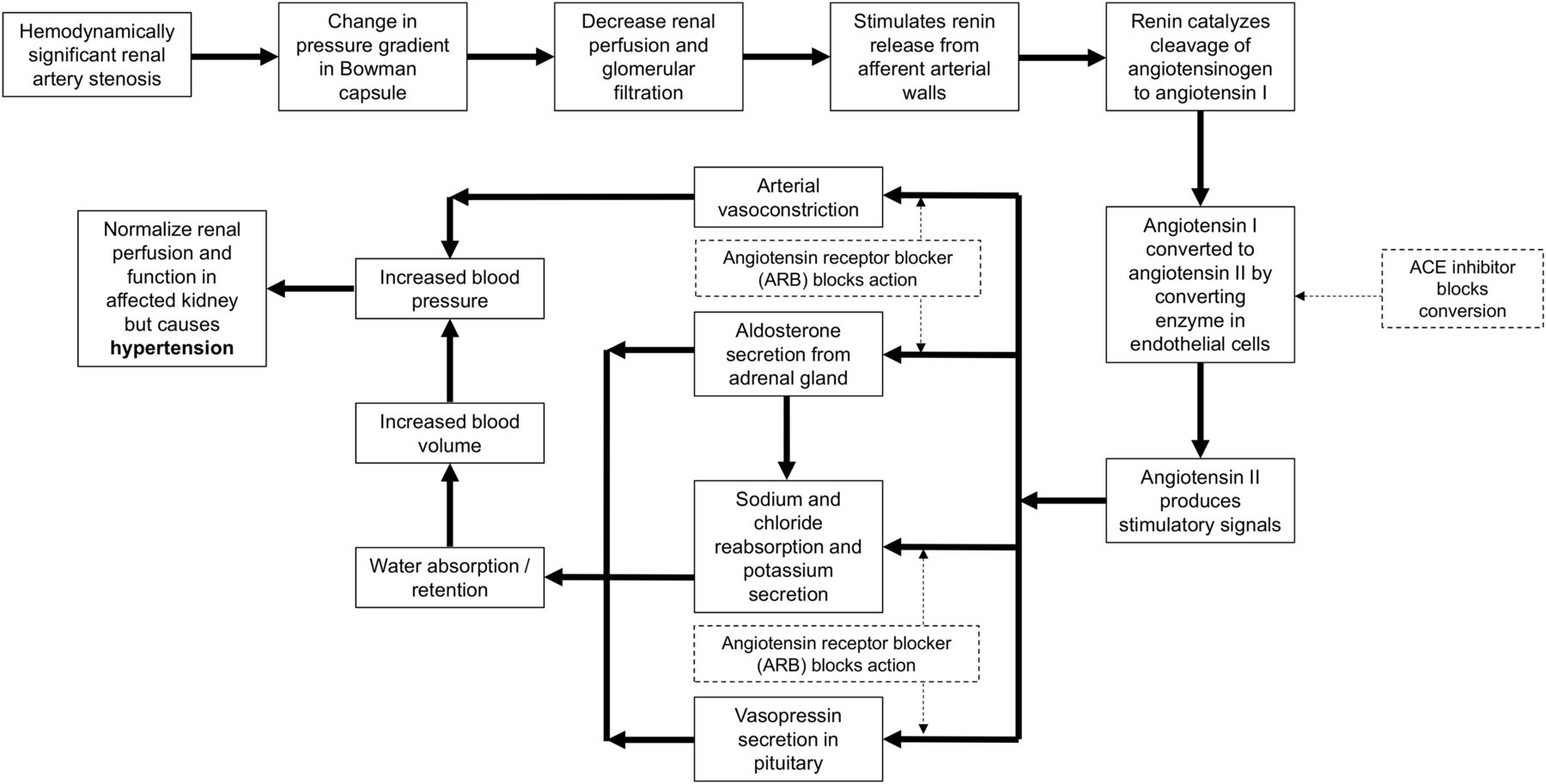
Flow chart of renin-angiotensin-aldosterone system response to decreased blood flow (solid lines). Inhibition of this response (dashed lines) can be undertaken by ACE inhibitors acting to prevent conversion of angiotensin I to angiotensin II or by ARBs antagonizing angiotensin receptor at numerous sites of action.
Captopril is an ACE inhibitor that competitively blocks the ACE to decrease blood pressure (1,2,8–12). Although captopril is the prototype ACE inhibitor, it differs from most other ACE inhibitors in that it is not an ester prodrug (9), thus allowing more rapid onset of action and shorter duration of effect, making it ideal for an interventional study. Prodrugs such as enalapril have slower onset and longer duration; therefore, therapeutically, they allow a once-per-day dosing regime (9).
Mode of Action
Captopril blocks the conversion of angiotensin I to angiotensin II (1,2,7–13,15). This action inhibits the compensatory vasoconstriction response in the efferent arterioles in the kidney, leading to decreased glomerulus filtration in a kidney with a hemodynamically significant renal artery stenosis (1,2,7,11,12,15).
Pharmacokinetics
Captopril has an oral bioavailability of 70%–75% (9,16). It has 30% plasma protein binding, 40%–50% is excreted unchanged in urine, and the remaining 50%–60% is metabolized in the liver (9,16). The elimination half-life is 2–3 h (9,16) but can be increased in renal dysfunction (16). After oral administration, onset of action occurs within 15 min, with peak activity seen at 45–70 min and significant effects lasting 6–12 h (1,9,16).
Usual Indications
Captopril is not the front-line ACE inhibitor but is used for treatment of hypertension in heart failure and after myocardial infarction (1,7–11,16). The properties that make it the preferred ACE inhibitor for renal imaging are the same properties that make it inferior to other ACE inhibitors for therapy. These include onset, half-life, and duration of effect.
Use in Nuclear Medicine
Captopril is used in conjunction with renal imaging (renogram) for the detection and characterization of the hemodynamic significance of renal artery stenosis in renovascular hypertension (1,2).
Proper Use and Dose Administration
Patients should discontinue ACE inhibitor therapy, including ARBs, before the study (1). The standard dose is 25 mg orally 60 min before commencement of the renal scan; however, 50 mg may be required if the requisite change in blood pressure does not occur (1,2,12,15). Patients should be monitored (blood pressure) for 60 min after receiving captopril and kept hydrated (1 L). Improved absorption and bioavailability can be achieved by crushing the tablet before administration (1).
Contraindications
Captopril is contraindicated when there is known hypersensitivity to captopril and in patients with angioedema, high renin levels, dehydration, salt depletion, recent dialysis, and aortic stenosis (9,12,16). Captopril use should be avoided during pregnancy (7,9,16).
Warnings and Precautions
Captopril should be used with caution in patients with scleroderma, lupus, depression, diabetes, and liver dysfunction (9). Therapeutically, captopril is not recommended in patients with renovascular disease such as renal artery stenosis; however, a single dose for an interventional procedure can be safely undertaken with patient supervision (9,16). Captopril is excreted in breast milk but is unlikely to be harmful to a nursing infant, with relative infant doses on the order of 0.02% of the dose to the mother (16). Captopril should be used with caution in the elderly or those with peripheral vascular disease (16).
Adverse Reactions
Single doses of captopril for interventional studies may elicit several adverse reactions, including hypotension, dizziness, tachycardia, chest pain, loss of taste, fever, and a rash (1,7–10,16). Longer-term therapy may lead to a dry cough, proteinuria, neutropenia, and thrombocythemia (1,7). The intractable dry cough associated with ACE inhibitors is well documented, has implications for therapeutic compliance, and drives some patients toward ARB therapy. The dry cough is thought to be due to incidental inhibition of the enzyme that breaks down bradykinin (9).
Common Interactions
Other ACE inhibitors and ARBs need to be withheld for 3 d before the captopril scan (15). This withholding need be only 1 d if the patient is being treated therapeutically with captopril. The patient may experience severe hypotension if captopril is administered concurrently with diuretics or antihypertensive medications (9,16). The most serious interaction for ACE inhibitors is hyperkalemia due to the additive effect of potassium retention with diuretics and NSAIDs (9). Potassium-sparing diuretics and any potassium supplements should be ceased before ACE inhibition therapy (9,16). Captopril can decrease lithium and digoxin excretion and increase the risk of toxicity for both (9,16). Antacids reduce the bioavailability of captopril (16).
Enalapril (Vasotec; Valeant International Bermuda)
General Information/Drug Class
Enalapril is not an ACE inhibitor itself but rather an ester prodrug that is enzymatically converted in the liver to the active metabolite enalaprilat (1,9,11). The principles, mode of action, usual indications, use in nuclear medicine, contraindications, warnings and precautions, adverse reactions, and common interactions are as per captopril, discussed above.
Pharmacokinetics
Enalapril has an oral bioavailability of 60% and is rapidly biotransformed to enalaprilat (9,16). Enalaprilat has 50%–60% plasma protein binding, 90% is excreted in urine, and the remaining 10% is excreted in feces (9,16). The elimination half-life is 11 h (9,16) but is multiphasic (16). After oral administration, onset of action occurs within 60 min, with peak activity seen at 4–6 h and significant effects lasting 24 h (1,9,16). The onset and peak times can be reduced to 15 min and 60 min, respectively, with intravenous administration (9,16).
Proper Use and Dose Administration
Enalapril can be administered by oral tablet or intravenous routes; however, the benefits of enalapril over captopril (more predictable action) are best demonstrated with an intravenous route (1). A dose of 0.05 mg/kg diluted in 10 mL of saline is injected intravenously slowly over 5 min (1,2,12). If the blood pressure drops more than 30% from baseline, the saline infusion is increased until blood pressure is back to the reference level (1). The renal scan can begin 10–15 min after enalapril in response to changes in blood pressure (1,12).
Adverse Reactions
Adverse effects from intravenous enalapril can be reduced using the slow intravenous infusion rate (5 min), and this is particularly helpful in reducing the risk of significant hypotension (1).
INTERVENTIONAL HEPATOBILIARY STUDIES
Interventional hepatobiliary scans are performed frequently in general nuclear medicine. Sincalide stimulation of the gallbladder and morphine augmentation are the principal interventional applications discussed here and summarized in Table 2. The less commonly used phenobarbital intervention in the jaundiced neonate is also discussed.
Sincalide (Kinevac; Bracco Diagnostics Inc.)
General Information/Drug Class
Cholecystokinin is an endogenous, 33-amino-acid polypeptide hormone secreted in the duodenum in response to a fatty meal (9,17). Sincalide is the exogenous active portion (octapeptide) of cholecystokinin (2,17).
Mode of Action
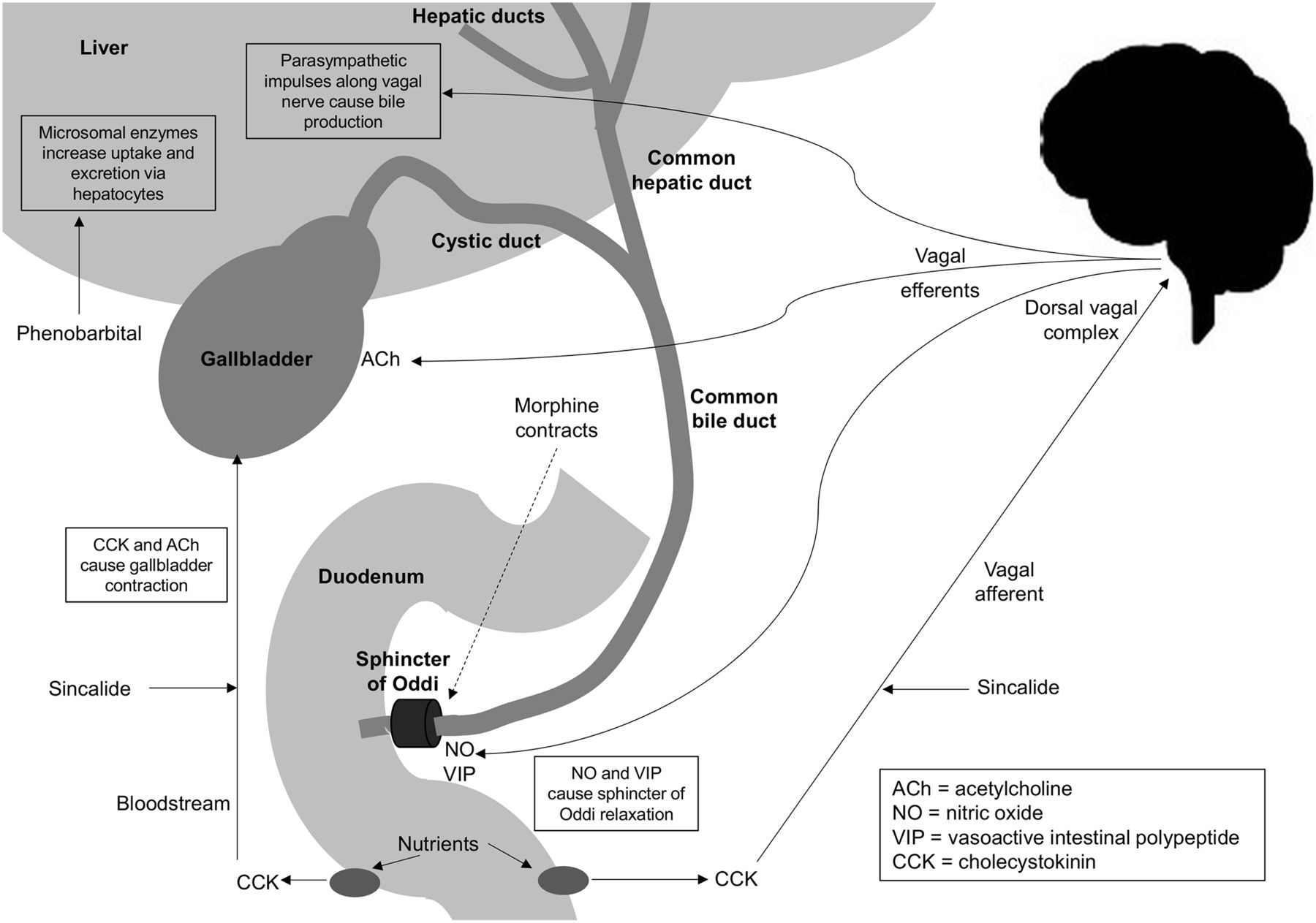
Sincalide is a synthetic peptide representing the active portion of the endogenous cholecystokinin hormone (2,12,15,17). Cholecystokinin/sincalide is released from the duodenal mucosa in response to a fat stimulus, which causes contraction of the gallbladder (17). Cholecystokinin/sincalide stimulates the dorsal vagus complex, which, through vagal efferents, relaxes the sphincter of Oddi, contracts the gallbladder, and stimulates bile production (Fig. 3). The primary mechanism for gallbladder contraction, however, is cholecystokinin/sincalide entering the bloodstream to act directly on the gallbladder. Gallbladder contraction occurs when the serum cholecystokinin/sincalide levels reach a threshold (2,14,15,17,18). Because gallbladder contraction only occurs when serum cholecystokinin/sincalide levels reach a threshold, intravenous introduction of a synthetic active portion is associated with 5 times higher potency (2,15,18).

Schematic representation of release of cholecystokinin to produce gallbladder contraction directly and via vagal afferents. Implications of exogenous sincalide are demonstrated. Relaxation (cholecystokinin) and contraction (morphine) effects on sphincter of Oddi are illustrated. Phenobarbital stimulation of biliary excretion independently of vagal nerve is also illustrated.
Pharmacokinetics
There are limited pharmacokinetic data available for sincalide. Sincalide has an intravenous bioavailability of 100% (9,16). After intravenous administration, onset of action occurs within 5–15 min, with peak activity seen at 40 min (1,9,16). The gallbladder should start emptying within 2 min of intravenous administration of sincalide and complete by 11 min (1). A 35% gallbladder ejection fraction or greater is considered normal (1,2).
Usual Indications
The usual use of sincalide is to contract the gallbladder as an adjunct to cholecystography (16). Sincalide has also been used as a function test of the pancreas in combination with secretin (16). Endogenous cholecystokinin has been used to manage obesity because cholecystokinin inhibits gastric motility and increases satiety (8,9).
Use in Nuclear Medicine
Sincalide is used to assess gallbladder function and the patency of the biliary system after gallbladder contraction, including calculation of the gallbladder ejection fraction (1,2,17,18). Sincalide intervention can enhance the specificity of the procedure and shorten the time to reporting the study (1,17,18).
Proper Use and Dose Administration
The patient should have fasted for a minimum of 4 h and a maximum of 6 h to ensure that gallbladder filling of the radiopharmaceutical is not impeded by residual endogenous cholecystokinin (45-min half-life) contracting the gallbladder or a full gallbladder due to absence of any endogenous cholecystokinin for a long period (1). The dose of sincalide is 0.02 μg/kg intravenously over 5 min (1,12,15,17,18). A larger second dose of 0.04 μg/kg diluted in 10 mL of saline administered intravenously over 5 min may be used if gallbladder contraction is not achieved with the first dose (1,12).
Contraindications
Sincalide is contraindicated in known hypersensitivity or intestinal obstruction (1,12). It is contraindicated in pregnancy because of the risk of spontaneous abortion.
Warnings and Precautions
Sincalide should not be administered after morphine (12).
Adverse Reactions
Adverse reactions are common but transient for sincalide and include abdominal pain and nausea (1,12,16). Less commonly patients may experience dizziness, flushing, and the urge to defecate (1,16). Adverse reactions can be minimized using a diluted dose (10 mL) and slow intravenous infusion (5 min) (1).
Common Interactions
There are no documented drug interactions, because of sincalide’s short-lived and mild action. Morphine and other opiates should, however, be stopped for 3 half-lives.
Morphine
General Information/Drug Class
Morphine is an opioid agonist that is mostly selective for μ-receptor (OP3) but can interact with other opioid receptors (κ and δ) at high doses (7–11,16,18). As an analgesic, the mechanism of action of opioids is not well understood, but inhibition of substance P release from neurons is a key aspect (8–11). Opioid receptors are G-protein–coupled transmembrane receptors that can be activated (agonists include morphine), inhibited (antagonists include naloxone), or partially agonized (e.g., buprenorphine) (8–11).
Mode of Action
Morphine increases smooth muscle tone, particularly the sphincters of the gastrointestinal and biliary tracts (8,16–18). Morphine contracts or constricts the sphincter of Oddi at the junction of the common bile duct and duodenum (Fig. 3), causing back pressure in the common bile duct (1,2,9,12,15,17,18). The back pressure in the common bile duct will allow a patent cystic duct to fill the gallbladder with bile and radiopharmaceutical (1,2,12,15,17,18). In doing so, this allows differentiation of chronic cholecystitis (incomplete cystic duct obstruction) from acute cholecystitis (complete cystic duct obstruction). It should be noted that for this intervention to be successful, there needs to be adequate clearance of the radiopharmaceutical from the liver after morphine administration. Newer radiopharmaceuticals with rapid clearance may require a second small (top-up) dose of radiopharmaceutical to avoid false-positive studies for acute cholecystitis.
Pharmacokinetics
Morphine can be administered via numerous routes, but for hepatobiliary augmentation, the dose is administered intravenously. Subanalgesic doses of 0.04 mg/kg intravenously over 3 min deliver 100% bioavailability. Morphine has 35% plasma protein binding; 50% is metabolized in the liver (conjugated with glucuronic acid) to morphine-3-glucuronide, which is inactive and excreted by the kidneys (16). A further 5%–15% is metabolized to morphine-6-glucuronide, which is more potent than morphine (1- to 2-h half-life) (8,9,16). Ten percent is excreted in the kidneys unchanged, and 10% is excreted as conjugates in bile and feces (16). The elimination half-life is 2 h (9,16), but morphine-3-glucuronide has an elimination half-life of 2.4–6.7 h (16). After intravenous administration, onset of action occurs within 5 min, with peak activity seen at 20 min and significant effects lasting 4–5 h (1,9,16). Morphine crosses the placenta and enters breast milk (16).
Usual Indications
Morphine is primarily a strong opioid analgesic (1,7,8,10,11) when given in therapeutic doses. Morphine is used for the relief of moderate to severe pain and to relieve anxiety associated with severe pain (7,8,10,11,16). It has been used as a hypnotic for insomnia due to pain (7,8,10,11,16).
Use in Nuclear Medicine
Morphine helps to differentiate chronic from acute cholecystitis. Morphine increases the specificity and shortens the duration of the biliary study in these patients (by avoiding delayed imaging) (1,2,17–19).
Proper Use and Dose Administration
If the gallbladder is not visualized 60 min after administration of the biliary radiopharmaceutical, but the common bile duct and duodenum are evident, morphine might be considered to avoid delayed imaging (1,2,12,17–20). Morphine has no role in the nonvisualized gallbladder in postcholecystectomy patients. If the duodenum is not visualized, morphine should not be administered (1). An intravenous administration over 1–3 min of 0.04 mg/kg is the standard dose, with imaging continuing for 30 min (1,2,17–20).
Contraindications
Although the doses being used in this interventional application are subanalgesic, morphine remains contraindicated in patients with a known allergy to morphine and respiratory depression (1,12,16,19). It should not be used in comatose patients (16).
Warnings and Precautions
Morphine should be used with caution in patients with seizures, head injuries, asthma, chronic lung disease, hypothyroidism, adrenocortical insufficiency, pheochromocytoma, kidney or liver dysfunction, prostatic hyperplasia, hypotension, shock, inflammatory or obstructive bowel disorders, myasthenia gravis, or pancreatitis (1,9,16). Caution should be used in cases of opioid addiction (16). Caution should also be exercised in pregnancy, and morphine is excreted in breast milk (16).
Adverse Reactions
Although severe adverse reactions are uncommon with subanalgesic doses, several adverse reactions can occur, including respiratory depression, dizziness, drowsiness, sedation, nausea, vomiting, constipation, and sweating (1,7–10,16,19). With longer-term use, patients may experience dry mouth, facial flushing, headache, vertigo, bradycardia, tachycardia, palpitations, orthostatic hypotension, hypothermia, restlessness, changes in mood, decreased libido, hallucinations, and miosis (7,8,16). Large doses of opioids produce respiratory depression and hypotension; circulatory failure and coma are possible (7,16). Morphine has a dose-related histamine-releasing effect (unlikely to be of significance in biliary scan doses) that can cause urticaria, pruritus, and hypotension (16).
Common Interactions
Therapeutic doses of narcotic analgesics need to be ceased for 5 half-lives to avoid interference with the biliary scan (15). Although morphine augmentation studies use subanalgesic doses, consideration should be given to potential interactions with other medications. Morphine interacts with alcohol and other central nervous system depressants to enhance respiratory and central nervous system depression and hypotension (9). Monoamine oxidase inhibitors can enhance the effects of morphine, and rifampicin can reduce the analgesic effects of morphine (9,16). There is an additive effect with benzodiazepines, and H2 histamine antagonists inhibit morphine metabolism (16). Clomipramine and amitriptyline increase the plasma availability of morphine (16). Naloxone is a narcotic antagonist and can be used with doses of 0.4 mg intravenously as an adjunctive medication in response to a severe allergic reaction to morphine (1).
Phenobarbital (Luminal; Bayer)
General Information/Drug Class
Phenobarbital is a long-acting barbiturate sedative and hypnotic that acts through γ-aminobutyric acid type A receptor agonism (positive modulation) (10,21). It also acts as an anticonvulsant (8,10,21).
Mode of Action
Phenobarbital enhances and accelerates radiopharmaceutical excretion by the biliary system, allowing differentiation of biliary atresia from neonatal hepatitis in the jaundiced neonate (15). Phenobarbital enhances the entire hepatic transport system independently of vagal induction (15). Phenobarbital induces microsomal enzymes in the liver, thus increasing uptake and excretion of several compounds, including bilirubin (Fig. 3) (1,12). Consequently, phenobarbital enhances biliary excretion (including biliary radiopharmaceuticals) if the biliary system is patent but not in biliary atresia (1,12).
Pharmacokinetics
Phenobarbital has significant variability with respect to pharmacokinetics. Phenobarbital has an oral bioavailability of 90% (16). It has 45%–60% plasma protein binding, 25% is excreted unchanged in urine, and the remainder is only partially metabolized in the liver (16). The elimination half-life is 75–120 h (16). After oral administration, onset of action occurs within 30–60 min, with peak activity seen at 2–12 h and significant effects lasting 4–48 h (1,16).
Usual Indications
Phenobarbital is used as an antiepileptic to control partial and generalized tonic clonic seizures (10,16). It can also be used in the emergency management of acute seizures (10,16).
Use in Nuclear Medicine
Phenobarbital is used to differentiate biliary atresia from neonatal hepatitis in the jaundiced neonate and to increase the sensitivity and specificity of the procedure (1,2).
Proper Use and Dose Administration
Phenobarbital is administered orally for 5 d before the biliary study at a dose rate of 5 mg/kg/d usually split into 2 doses (i.e., 2.5 mg/kg twice daily for 5 d) (1,2).
Contraindications
Phenobarbital is contraindicated where there is a known allergy to it and in patients with severe respiratory depression (1,2,16).
Warnings and Precautions
Caution should be exercised in children and the elderly, patients with acute pain, and patients with depression (16). Caution is also required for impaired liver, kidney, or lung function (16).
Adverse Reactions
Although sedation is the most common adverse reaction, phenobarbital can produce several common side effects, including respiratory depression, drowsiness, lethargy, rash, nausea, vomiting, and a paradoxic hyperexcitement in children (1,8,10,12,16). Mood changes and depression can be seen with longer-term use (16). Nystagmus and ataxia might occur at high doses (16). Hypersensitivity is uncommon but can occur (16).
Common Interactions
By virtue of the role of phenobarbital in activating microsomal enzymes in the liver, it can have a significant negative effect on drugs metabolized in the liver, including analgesics, antibacterials, antiarrythmics, antidepressants, antiepileptics, antipsychotics, antivirals, β-blockers, calcium channel blockers, digoxin, ciclosporin, diuretics, theophylline, and some vaccines (16). Phenobarbital effects can be enhanced by central nervous system depressants and alcohol (16).
CONCLUSION
An understanding of basic pharmacology for renal and biliary interventional medications allows enhanced practice. Specifically, this deeper understanding of pharmacology, indications, contraindications, warnings, precautions, proper use, drug interactions, and adverse reactions for each medication to be used ensures that nuclear medicine technologists meet the minimum capabilities for their scope of practice (4). This, in turn, translates to safer practice and better patient outcomes.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Aug. 3, 2018.
CE credit: For CE credit, you can access the test for this article, as well as additional JNMT CE tests, online at https://www.snmmilearningcenter.org. Complete the test online no later than December 2021. Your online test will be scored immediately. You may make 3 attempts to pass the test and must answer 80% of the questions correctly to receive 1.0 CEH (Continuing Education Hour) credit. SNMMI members will have their CEH credit added to their VOICE transcript automatically; nonmembers will be able to print out a CE certificate upon successfully completing the test. The online test is free to SNMMI members; nonmembers must pay $15.00 by credit card when logging onto the website to take the test.
REFERENCES
- Received for publication May 23, 2018.
- Accepted for publication July 16, 2018.




{kind=link}
{kind=link}
{kind=link}