


Abstract
The intention of this review is to provide information about the rapidly evolving field of molecular imaging and its potential impact on the clinical practice of nuclear medicine. On completing this article the reader should be able to define molecular imaging, describe the ways in which molecular imaging can be used, identify some of the biologic processes that can be targeted with molecular imaging agents, and list the modalities that can be used for molecular imaging, along with the strengths and weaknesses of each.
Molecular imaging has become something of a buzzword in recent years and often is portrayed as a key component of personalized medicine, itself another popular buzzword. The Society of Nuclear Medicine (SNM) and its Molecular Imaging Center of Excellence (MICoE) have adopted an official definition of molecular imaging (1): “Molecular imaging is the visualization, characterization, and measurement of biological processes at the molecular and cellular levels in humans and other living systems.” The objective of this article is to elucidate what molecular imaging is, how it is done, and how it is used both clinically and in research. Molecular imaging comprises a range of techniques, spanning not only several imaging modalities but also many diseases and organ sites. Many of the examples presented in what follows focus on cancer, primarily because that is the research area in which the authors are most heavily involved.
HOW CAN MOLECULAR IMAGING BE USED?
Traditionally, the primary role of medical imaging has been to aid medical diagnosis through the visualization of the presence, location, and extent of pathologies. Because molecular imaging techniques are capable of providing functional information at the cellular level, they offer the potential to move beyond mere identification and localization of diseased tissues to the characterization of the molecular processes involved.
The ability to characterize disease at the molecular level via imaging provides a powerful tool for clinical treatment planning and in the characterization of novel therapeutic regimens in the preclinical setting (clinical trial modeling). Increasingly, therapeutic strategies for oncology use pharmaceuticals that directly inhibit the activity of specific molecular pathways. For example, there are currently numerous drugs in the clinic that are designed to inhibit epidermal growth factor receptor (EGFR), human epidermal growth factor receptor-type 2 (HER-2), and vascular endothelial growth factor receptor (VEGFR) signaling. These agents are frequently used as single agents or in combination with other molecularly targeted therapies or standard chemotherapy. The current clinical emphasis on molecularly targeted therapeutics underscores a key objective of personalized medicine: pairing the most appropriate treatment with each patient on an individualized basis. However, the enormous complexity of using multiagent, molecularly targeted therapeutic regimens to treat cancer increases the already considerable pressure on those running clinical trials and the pharmaceutical industry to develop and validate efficient and robust biomarkers for assaying the clinical and biologic activity of these interventions. Molecular imaging can play a key role in this area.
Again using cancer as the example, knowing something about the molecular characteristics of the tumor before starting treatment decreases the chances that an ineffective therapy will be used. Molecular imaging can be used as a means of stratifying patients into those likely to respond to a molecularly targeted therapy from those likely to not respond, such as identifying HER-2 overexpressing breast cancers for treatment with trastuzumab (Herceptin) (2). Similarly, baseline tumor 18F-fluoroestradiol (18F-FES) uptake, a validated measure of estrogen receptor expression (3), and metabolic flare assessed by 18F-FDG PET after estradiol challenge are both predictive of responsiveness to endocrine therapy in estrogen receptor–positive breast cancer (4). By individualizing treatment plans in this way, the hope is that patient outcomes will be improved while at the same time health care costs are controlled through better use of resources. Although in certain cases such molecular information can be obtained through biopsy, there are many tumor sites, such as the liver and brain, for which biopsies are sufficiently risky that there is a clear preference for a less invasive readout. Furthermore, sampling errors can occur in biopsies, and there also can be discordance in expression profiles between primary and metastatic tumor sites.
Another area of patient care in which molecular imaging has great potential is in the evaluation of treatment response. The current standard method for using imaging to assess treatment response in clinical cancer trials is the application of the Response Evaluation Criteria in Solid Tumors. This approach is based on the determination of the largest linear dimension of a tumor as assessed by CT or MRI. Research has shown, however, that tumor shrinkage exhibits a considerable lag time after therapy, being preceded by changes in metabolism and cellular proliferation, for example, which can be detected through molecular imaging. Molecular imaging techniques also can be used to test whether specific pathways targeted by therapies have indeed been altered, such as probing angiogenesis via expression of the VEGFR.
Looking beyond the clinic, molecular imaging has become a key component of the modern drug-development process. A major driver of the enormous costs of bringing a new drug to market is failure in the late stages of the clinical trial process, and many pharmaceutical firms now incorporate molecular imaging with the objective of lowering these costs through earlier identification of drugs that are not only likely to be successful but also, more important, likely to fail, because halting those trials earlier leads to significant cost savings. There are several ways in which molecular imaging can be incorporated into drug development. One way is through the direct labeling of the novel drug to turn it into a molecular imaging agent. This approach provides a useful means of assessing the pharmacokinetics of the drug. The ability to obtain biodistribution information using tracer levels of the drug, sometimes referred to as microdosing, can provide insight into potential toxicity concerns while at the same time helping to verify that the drug reaches its target.
Another approach for investigating the properties of new drugs with molecular imaging is to use a radiotracer that shares a pathway or target with the drug in question as a means of determining the appropriate dose and dosing schedule. Through kinetic modeling, it is possible to determine the occupancy of the drug at the target site based on the degree of binding of the molecular imaging agent. Such studies use molecular imaging to probe the relationship between receptor occupancy levels of the drug under investigation and clinically observable pharmacologic effects at a given dose. These data can be used to inform whether a drug under investigation has a sufficient safety margin with respect to possible toxicity or side effects at an efficacious dose.
Another area of drug development in which molecular imaging can play an important role is as a biomarker in clinical trials. The basic concept of the biomarker is to use some measurable quantity as an indicator of a biologic process and its response to treatment. An example of such a biomarker is the use of cholesterol levels as a measure of risk for coronary artery disease. The advantage of biomarkers in drug development is that they can significantly shorten the length of time required to complete a clinical trial, because in many cases the recognized endpoints, such as 5-y survival rate in cancer treatments, make the process slow and expensive. The SNM Clinical Trials Network was established recently to facilitate the incorporation of imaging biomarkers into multicenter clinical trials. In particular, this network will help in coordinating the production of the molecular imaging agents and standardization of the imaging protocols across multiple sites to streamline the inclusion of molecular imaging in the development of new therapeutics. A key task in such studies is to validate that the imaging readout serves as a biomarker. However, once a particular biomarker has been validated it can be used in clinical trials for any therapies that share the same outcome for which that biomarker serves as surrogate.
Moving beyond drug development, molecular imaging currently plays an important role in a great deal of biomedical research. The ability to study cellular and molecular processes using these minimally invasive techniques can provide insight into the mechanisms of disease onset and progression. The coupling of molecular imaging with reporter genes and transgenic mouse models of human diseases enables scientists to probe molecular pathways in ways that can not only reveal the fundamental processes that characterize these diseases but also identify possible targets for diagnosis and treatment. Although it remains to be seen whether molecular imaging will reach its full potential in the clinical realm, it already is well established as a critical tool in basic and translational research.
MOLECULAR IMAGING MODALITIES
Although molecular imaging as an identifiable field of research is a fairly recent phenomenon, it turns out that much of nuclear medicine as it is practiced can be considered molecular imaging. Those radiotracers that target specific receptors (octreotide), transporters (N-3-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl)tropane, or molecular processes (18F-FDG) are considered molecular imaging agents. On first glance, then, the nuclear medicine practitioner may be left wondering what the big deal is. The relationship between nuclear medicine and molecular imaging is not a one-to-one match, however, because there are nuclear medicine procedures that are not molecular imaging, whereas molecular imaging techniques can be applied using imaging modalities other than PET and SPECT. An example of the former would be the measurement of pulmonary perfusion with 99mTc-macroaggregated albumin, for which the radiotracer does not have a specific molecular target but rather is designed to stay within the vasculature. We present examples of the latter in the form of brief descriptions of nonnuclear molecular imaging approaches. Table 1 provides an overview and rough comparisons of the major molecular imaging modalities.
Comparison of Molecular Imaging Modalities
Although MRI is generally thought of as offering anatomic information, there are several methods by which physiologic and functional information can be obtained. An example of a physiologic readout is blood flow assessed through dynamic contrast-enhanced MRI. Molecular imaging with MRI typically involves contrast agents that incorporate gadolinium or iron oxide in the form of superparamagnetic iron oxide (SPIO) or ultrasmall superparamagnetic iron oxide (USPIO). These materials alter the relaxation times of nearby water protons (gadolinium) or perturb the local magnetic field (iron oxide), thereby influencing the MRI signal. Although MRI techniques can offer good spatial resolution (around 1 mm for clinical applications and down to 0.1 mm in animal studies), a major drawback of molecular imaging using MRI is that the sensitivity (minimum detectable concentration of agent) is typically several orders of magnitude lower than that of standard nuclear imaging methods, which means that any receptors targeted by the contrast agent must be available in large numbers. Another difficulty in using MRI for molecular imaging is that quantitative measurements cannot be easily made. In nuclear medicine, the signal is generated solely from the radiotracer, whereas the signal in MRI comes from a vast number of hydrogen protons, with the contrast agent altering the signal properties of some of these protons. Absolute quantification requires knowledge of both how the contrast agent alters signal and what the signal would have been in the absence of the contrast agent.
Another molecular imaging method that is closely related to MRI is magnetic resonance spectroscopy (MRS). MRS can be used to measure the relative abundance of endogenous compounds, removing the need for contrast agents entirely. MRS relies on the fact that the resonant frequency of a specific nuclear species (i.e., hydrogen protons) depends on its molecular environment, an effect known as the chemical shift. By measuring the strength of the MRI signal while scanning across a suitable range of frequencies, it is possible to measure the relative abundance of certain molecules. When such measurements are conducted with appropriate pulse sequences, these signals can be confined to a specific region of the body and repeated measurements used to create spatial maps of molecular abundances (this is sometimes referred to as MRS imaging), although typically at much coarser spatial resolution than standard MRI. MRS measurements of major metabolites in the brain such as N-acetyl aspartate and choline can be useful in characterizing brain tumors (5).
There are other nuclei besides that of 1H that possess magnetic moments suitable for use in MRI or MRS. Some of these, for example, 13C and 19F, are of sufficiently low natural abundance in the body that, when used as a label on an exogenous contrast agent, the in vivo distribution of that agent can be measured with minimal background. Although this approach is similar to the nuclear imaging of radiotracers, it suffers from lower sensitivity because the net magnetization that creates the signal arises from the roughly 1 part in 100,000 asymmetry in the populations of the energy levels (parallel and antiparallel spin states). The sensitivity can be increased through the use of hyperpolarization to create a less even distribution of energy levels, thereby enhancing the magnetization. This hyperpolarization must be created outside the body, and in the case of 13C it has been used to label metabolic substrates, such as pyruvate, before injection (6). Through MRS, it is then possible to measure the rate of metabolic turnover, for example, the conversion of pyruvate to lactate, because the 13C signal depends on the chemical state. Although such approaches offer the potential to measure biochemical pathways in exquisite detail, the short lifetime of the magnetization enhancement from the hyperpolarization (tens of seconds or less) makes this challenging.
An important recent development in ultrasound imaging has been the use of microbubbles as a contrast agent. These microbubbles are gas-filled (often perfluorocarbon) lipid shells that are highly echogenic because of the large acoustic impedance mismatch between blood and tissue and gas. The major application of microbubbles has been as a vascular contrast agent to assess blood flow and perfusion (7). More recently, several investigators have turned microbubbles into molecular imaging agents by functionalizing the bubbles through the addition of ligands to the shells (8). Because the microbubbles are relatively large (microns), their use is limited to vascular targets, such as the EGFR. To acquire these images, the microbubbles are injected intravenously and the ultrasound transducer is positioned to image microbubbles as they appear in the body region of interest. The real-time nature of ultrasound acquisition enables visualization of the delivery, accumulation, and washout of the microbubbles. Periodic ultrasound pulses of higher power can be used to collapse the bubbles within the field of view, allowing the uptake and binding of the microbubbles in that region to be imaged multiple times and potentially quantified.
Probably the most widely used imaging modality for preclinical molecular imaging is optical imaging. The optical imaging paradigm is attractive for preclinical studies for numerous reasons, including throughput, sensitivity, ease of use, and overall cost. Optical imaging in vivo covers a range of techniques but can be broken down at the highest level into fluorescence and bioluminescence imaging. Bioluminescence and fluorescence imaging are theoretically capable of similar levels of sensitivity, yet in practice, bioluminescence imaging typically exhibits a considerable sensitivity advantage (several hundreds to a few thousand cells) over fluorescence imaging (tens of thousands of cells) due its fundamental lack of background emission. Signal detection in all varieties of optical imaging is most commonly accomplished using a lens-coupled charge-coupled device.
Bioluminescence imaging (BLI) is based on a biochemical reaction in which optical photons are created—the exact same process, in fact, by which fireflies create their characteristic glow on summer nights. Using molecular biology techniques, researchers have isolated the gene (luciferase) responsible for this reaction and have been able to genetically engineer mammalian cells, such as tumor cells, to express this gene. A standard BLI study involves injecting luciferin, the substrate required for the reaction, into a subject in which luciferase-expressing cells are present. When luciferin enters a cell in which the luciferase enzyme is present, a chemical reaction involving enzyme, substrate, adenosine triphosphate, and oxygen produces a detectable photon. BLI provides a sensitive means of detecting the presence and location of the luciferase-expressing cells, making it useful for studying such things as tumor growth and metastases. An extension of this technique involves cloning the luciferase gene with the promoter region of another gene of interest. The amount of luciferase activity is then related to the expression level of the gene under study, enabling researchers to study temporal and environmental factors influencing gene expression. One example of this approach is the creation of transgenic mice expressing luciferase under the control of the mouse insulin promoter, enabling investigators to monitor β-cell function in models of diabetes using BLI (9). Although BLI is unlikely ever to find a clinical application, it is a powerful preclinical tool that is having an enormous impact on many areas of research.
Fluorescence imaging can be performed using dyes, quantum dots, and even proteins (the 2008 Nobel Prize in Chemistry was awarded to Chalfie, Shimomura, and Tsien for the development of green fluorescent protein for biomedical research). In all cases, the image acquisition first involves the excitation of the agent with an external light source at the appropriate wavelength, followed by the detection of the resulting photon emissions from the decay of the excited states. Like nuclear medicine, optical imaging can have a low background, but it has the added advantages of not involving ionizing radiation and offering the possibility of repeatedly obtaining signal from each molecule. The fluorescent (emissive) state can be accessed multiple times (although not infinitely so, because of a process known as photobleaching), making it possible for an individual group of molecules to contribute to multiple images over the course of a study, in contrast to radiotracers in which each probe can contribute (at most) only once to an image when its radionuclide decays. Signal generation is tied to the application of an external light source, and the lifetime of the excited states is quite short, meaning that the temporal resolution is quite good. The imaging instruments themselves are simple to use, and images can be acquired rapidly.
The major drawback to optical imaging arises from the strong absorption and scattering of photons at that wavelength scale. These effects limit the depth of penetration; result in modest, depth-dependent spatial resolution; and complicate quantification. The attenuation problem is particularly complicated because it affects both the excitation of the signaling molecules and their emissions. Many researchers are working on methods for creating tomographic images using optical probes, but the fact that nearly all of the detected photons undergo multiple scattering within the object makes the reconstruction process an ill-posed inverse problem for which unique solutions are difficult to obtain.
Although molecular imaging using optical methods will not be broadly applicable to clinical imaging because of the strong attenuation of optical photon wavelengths in tissue, there are some possible areas in which it may be viable. It may be possible to use fluorescent agents for imaging superficial sites, perhaps in identifying melanoma. Another arena in which such agents could be put to use is in surgery, particularly in the resection of tumors. A tumor-specific ligand labeled with a fluorescent dye could be injected before the surgery. Fluorescent imaging could then be performed during the resection to identify tumor cells to ensure that none was left behind. One other possible area of clinical application for optical imaging involves coupling fluorescent probes with endoscopy. An example here would be the detection of colorectal cancer, in which the use of an endoscope eliminates the limitation of depth of penetration of the fluorescence photons (10).
The specificity of targeted molecular imaging agents can make the interpretation of the images challenging. The benefit of anatomic information for image interpretation has led to the rapid adoption of so-called hybrid imaging devices, combining PET or SPECT with CT, for example. An added benefit of CT in these cases is the ability to use the anatomic information in carrying out attenuation and scatter correction, resulting in improved quantification. There is great interest in the molecular imaging community in multimodality imaging approaches that extend beyond the addition of anatomic information. Combining molecular imaging readouts (e.g., assessing glucose metabolism via 18F-FDG and apoptosis via 99mTc-annexin-V) provides a fuller picture of the disease state or its response to treatment. The emergence of hybrid PET/MRI systems will usher in new opportunities not only for integrating anatomic and molecular information but also for adding functional and physiologic information to the mix. This enhanced integration may allow, for instance, the physiologic response to a drug challenge to be monitored in concert with the displacement of a radiotracer by that drug.
MOLECULAR IMAGING AGENTS
Attributes Common to All Molecular Imaging Agents
In the most general sense, all molecular imaging agents consist of the same basic features regardless of the imaging modality with which they are used. By definition, a molecular imaging agent targets a specific molecular entity or process and therefore must contain a targeting moiety or carrier. This is the portion of the molecular imaging agent that is responsible for directing the probe to the proper target. Additionally, all molecular imaging agents contain a signaling moiety or sensor. The signaling moiety is the actual species that is responsible for producing the signal that is detected by the imaging system. Common signaling moieties include radionuclides such as 18F or 99mTc, fluorochromes, and microbubbles. In some cases, the targeting moiety can be directly labeled with signaling moieties, such as 18F labeling of 3′-deoxy-3′-fluorothymidine (18F-FLT) (Fig. 1A). In other cases, it may be advantageous to separate the signaling moiety from the targeting moiety using a synthetic spacer or linker (Fig. 1B). This may be particularly important if the labeling of the targeting species may in some way influence the binding of the imaging probe to the target. In either case, significant synthetic chemistry efforts are generally required to optimize the biologic and physical properties of a molecule intended to serve as a molecular imaging agent.
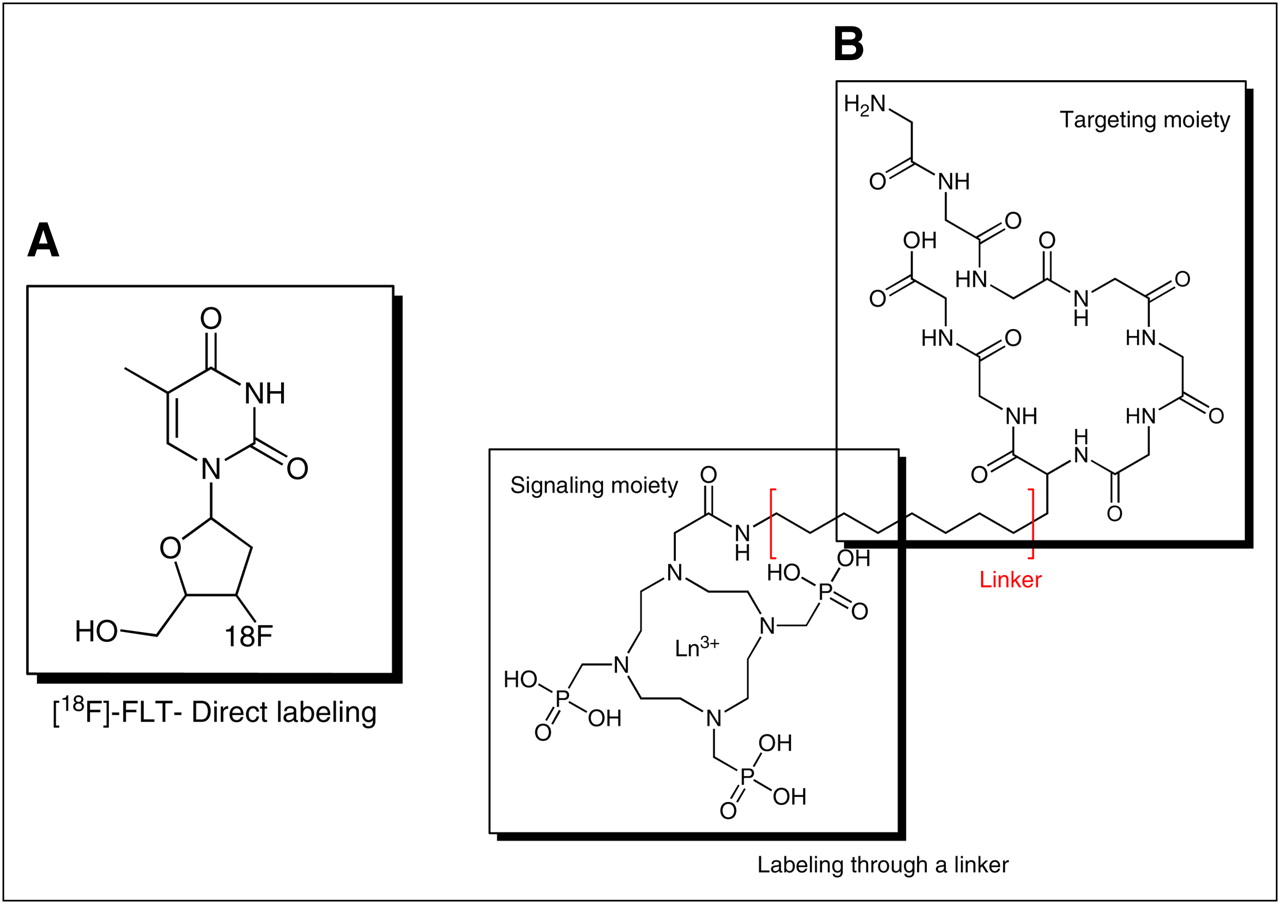
Examples of synthetic approaches to preparing molecular imaging agents: direct labeling (A) and labeling through linker (B).
It is important to emphasize that a specific targeting vector can be prepared by labeling it with any one of several different signaling moieties, or possibly more than one signaling moiety (11), creating the option of imaging the same target with different modalities. A good example of this are imaging probes based on annexin-V, which is commonly labeled with 99mTc for SPECT or near-infrared (NIR) fluorochromes for optical imaging but can also be easily labeled with 68Ga or 64Cu for PET. Flexibility in probe chemistry is attractive because it enables the users to easily tailor the imaging experiment and modality for specific purposes.
Biologic Processes Measurable with Molecular Imaging Agents
Advancement of the molecular imaging field is driven by the development of improved imaging hardware for use in the preclinical and clinical settings; identification and validation of new, biologically relevant imaging targets; and development of improved imaging probes derived from novel chemistries. Of these 3 essential facets that comprise most current molecular imaging research, hardware development and novel target discovery significantly outpace the development and clinical advancement of new molecular imaging probes, particularly with respect to cancer imaging.
To date, several imaging probes have been described that aim to measure fundamental biologic processes known to be dysregulated in diseased tissues, such as tumors and other diseases, including glucose use, proliferation, apoptosis, hypoxia, and angiogenesis (12). Many of these imaging probes have been used fairly extensively in the preclinical setting and some to a lesser extent clinically, but in reality, the most molecular imaging in the clinic consists of assessment of glucose use using 18F-FDG-PET. Because regulation of glycolysis is a complicated process involving several biologic factors capable of influencing the overall glycolytic pathway, 18F-FDG PET studies must be interpreted with some caution; imaging results can vary significantly depending on the extent and type of disease and cellular responses to a therapeutic intervention. Given the current emphasis placed on the development of sophisticated molecularly targeted and individualized therapeutic regimens for the treatment of cancer, expanding the imaging repertoire of the oncologist to include probes capable of reporting more specific molecular events and relevant downstream cellular physiology may be of considerable clinical importance. For these reasons, there is significant interest in the development and validation of additional novel imaging probes that have the potential to contribute insights into the molecular biology of disease and therapeutic response that may extend beyond what can be learned with currently existing tracers such as 18F-FDG. We will focus our discussion of contemporary molecular imaging probes around several facets of relevant biology.
Metabolism.
By far, the most commonly used PET tracer for clinical molecular imaging is 18F-FDG in oncology. 18F-FDG PET exploits the typically increased glucose metabolism of tumor tissues, compared with surrounding normal tissues. 18F-FDG is transported into tumor cells via glucose transport proteins (such as GLUT1), which tend to be upregulated in tumor cells. Once 18F-FDG is internalized, the tracer is phosphorylated to FDG-6-phosphate by an enzyme known as hexokinase. Unlike glucose-6 phosphate, FDG-6-phosphate does not enter glycolysis because of the presence of fluorine substitution at the 2- position, and the tracer becomes metabolically trapped. Subsequent accumulation of 18F-FDG in metabolically active cells leads to imaging contrast. Some key drawbacks of 18F-FDG include nonspecific accumulation in inflammation and high background accumulation in highly metabolic tissues such as muscle and normal brain.
As an alternative to assessing glucose metabolism with 18F-FDG, amino acid–based tracers can be used to measure protein metabolism. Tracers such as O-(2-18F-fluoroethyl)-l-tyrosine have been used with particular success in brain tumors (13) because, unlike 18F-FDG, these agents have little uptake in normal brain tissues. Amino acid–based tracers typically show little uptake in inflammatory lesions, making them an attractive alternative to 18F-FDG in some cases as well. A drawback of amino acid tracers is rapid metabolism and attendant radioactive metabolites present in blood and tissues, which can confound the interpretation of imaging data.
Proliferation.
Noninvasive imaging approaches designed to longitudinally assess cellular proliferation potentially offer considerable advantages over invasive approaches that rely on serial biopsy. For this reason, investigation into suitable methodologies for imaging proliferation has been undertaken by several groups. Historically, several PET tracers that are precursors for DNA synthesis have been explored and include 11C- and 18F-labeled nucleosides and structural analogs (14–16). One of the most promising nucleoside-based imaging probes described thus far has been 3′-deoxy-3′-18F-fluorothymidine (18F-FLT) (17–23). Theoretically, 18F-FLT and other nucleoside-based tracers serve as surrogate markers of proliferation by reporting the activity of the thymidine salvage pathway, a cellular mechanism that uses uptake of deoxyribonucleosides from the extracellular environment to provide dividing cells with DNA precursors. Like other nucleosides such as thymidine, cytidine, and guanosine, 18F-FLT is thought to be transported across the cell membrane by facilitated diffusion via low-affinity, nonconcentrative nucleoside carrier proteins that are conserved across nearly all animal cells (24). On cellular internalization, 18F-FLT is monophosphorylated in a reaction catalyzed by the cytosolic enzyme thymidine kinase 1 (TK1). Unlike thymidine, 18F-FLT is not readily incorporated into DNA (25), yet phosphorylation to 18F-FLT monophosphate results in intracellular trapping and subsequent accumulation. In many tissues, TK1 activity is regulated at transcriptional, translational, and posttranslational levels (26), and activity tends to be closely correlated with the DNA synthesis phase of proliferating cells (typically late G1 through S). However, TK1 activity is typically diminished in quiescent, nonproliferating cells (17,18,27,28). Since the late 1990s, many preclinical and clinical studies have been published exploring the utility of 18F-FLT PET as a quantitative metric to assess cellular proliferation in various species, tumor types, and organ sites (18,20–22,27,29) and, although it is not yet Food and Drug Administration-approved, use of this tracer is becoming more common clinically.
Cellular proliferation can also be estimated by measuring phospholipid metabolism with 11C-choline. Choline imaging has been shown to be particularly useful in brain cancer and prostate cancer imaging, where the increased activities of choline transporters and choline kinase are associated with increased cell membrane synthesis and proliferation. Similarly, 11C-acetate, a precursor of fatty acid synthesis, can be used as a surrogate marker of metabolism and proliferation and has shown utility in prostate cancer and other diseases.
Hypoxia.
Solid tumors larger than a few millimeters in diameter rapidly outgrow their nutrient and oxygen supply. Poorly oxygenated tumor tissues rapidly become hypoxic. Hypoxia is known to play a role in angiogenesis, metastasis, and resistance to therapy. For this reason, molecular imaging assessment of hypoxia potentially could be of considerable importance. Two PET tracers, 18F-fluoromisonidazole (30) and 64Cu-diacetyl-bis(N4-methylthiosemicarbazone) (31), have been evaluated rather extensively in various solid tumors. Both tracers have shown utility for measuring hypoxia, though additional validation studies are required and are under way at several institutions.
Apoptosis.
In normal cells, programmed cell death (or apoptosis) is a tightly regulated intracellular suicide program that is widely used as a method for shedding redundant or dysfunctional cells. Cellular regulation of apoptosis proceeds via a complex cascade of intracellular signaling machinery that is conserved across a majority of animal cells. Specific molecular defects in apoptosis regulation are closely associated with numerous diseases including neurodegenerative disease and cancer. Considerable research has been undertaken to more closely understand apoptosis, particularly with respect to the adaptive mechanisms that many tumor cells use to maximize survival. Indeed, an impaired apoptosis program is a feature of many types of malignant tumor cells. Though the term apoptosis itself was coined nearly 40 y ago by Kerr et al. (32) and an awareness of the general phenomenon was known for many years before that (33), numerous biologic factors affecting the molecular regulation of apoptosis still require further characterization at the basic science level.
Significant efforts have gone into the development of noninvasive imaging methods to longitudinally assess apoptosis. Many of these efforts have focused on the use of annexin-V, an endogenous, 36-kDa human protein that binds phosphatidyl serine (PS) with nanomolar affinity. Under most normal circumstances, PS is restricted to the inner leaflet of the cell membrane lipid-bilayer. However, the early execution phase of apoptosis is closely associated with the redistribution and externalization of PS to the cell surface. This fact forms the basis of targeting PS with imaging probes as a metric to assess apoptosis.
Numerous clinical studies have demonstrated promising results with SPECT of 99mTc-labeled annexin-V to assess apoptosis in patients with cancer (34–39), myocardial infarction (40,41), ischemic preconditioning (42), vulnerable atherosclerotic plaques (43), acute stroke (44,45), and Alzheimer dementia (46). Additionally, fluorophore-labeled versions of annexin-V have been used for flow cytometry and for imaging response to therapy in animal models (47,48). For example, we recently reported use of an annexin-V derivative labeled with a NIR dye to assess response to cetuximab therapy in mouse models of colorectal cancer (49). A limitation of measuring apoptosis as a treatment response outcome is that there are several other nonapoptotic forms of cell death such as necrosis, autophagy, and mitotic catastrophe. Each of these nonapoptotic cell death mechanisms is inducible by therapeutics, with the consequence that imaging of apoptosis does not necessarily provide a complete picture of the effectiveness of a therapy in inducing cell death.
More exploratory methods to image apoptosis that may become more common in the future include the assessment of caspase activity using peptides or small molecules and the targeting of activity of cell surface death receptors.
Receptors.
A key area of molecular imaging research has historically been and continues to be based on imaging agents that bind to cell surface receptors with high specificity. PET, with its high sensitivity and quantitative capability, is particularly powerful for such studies, especially those performed in the brain. Neuroreceptor imaging plays an important role in the diagnosis and study of neurodegenerative disorders such as Parkinson disease. Another major use of neuroreceptor imaging involves quantitative occupancy studies central to drug discovery for the treatment of psychiatric disorders. Additionally, many studies are conducted to evaluate the effects of drugs, including drugs of abuse such as methamphetamine and cocaine, on brain function (50). One of the more common targets of such neuroreceptor imaging studies has been the dopaminergic system, and numerous tracers such as 18F-fallypride (51) and 11C-raclopride (52) exist for this purpose.
Molecular imaging of receptor expression is also important in oncology. In breast cancer, 18F-FES has been shown to predict those patients who will respond to endocrine therapy (3,53). Additionally, imaging probes targeting EGFR and HER-2 have been prepared and shown to have potential for the evaluation of receptor status in tumors (49,54). 111In-diethylenetriaminepentaacetic acid-d-phe-octreotide is a routine clinical tool for the evaluation of somatostatin receptor in neuroendocrine tumors, yet PET analogs bearing either 68Ga or 64Cu are also being evaluated to extend somatostatin receptor imaging to PET, where sensitivity is somewhat better.
Angiogenesis.
Angiogenesis, or the formation and recruitment of new vasculature, is a highly orchestrated biologic process that in healthy individuals is primarily confined to wound healing and reproduction. Dysregulated angiogenesis is a pathologic condition and characteristic of several common diseases including diabetes, psoriasis, rheumatoid arthritis, and cancer (55). Angiogenesis plays a central role in the development and progression of tumors, as neovascularization is required to supply oxygen and nutrients to rapidly growing tumor cells and in turn facilitates the spread of metastases (56). Tumor-induced angiogenesis is predominately driven by paracrine vascular endothelial growth factor (VEGF) signaling between tumor or stromal cells, which can secrete a variety of soluble VEGF ligands, and endothelial cells, which express tyrosine kinase VEGFRs (57–59).
The VEGF family of receptors is an attractive class of imaging targets because they are primarily expressed on the surface of endothelial cells (60), enabling facile delivery of imaging compounds throughout the bloodstream. PET and SPECT and optical imaging methods, such as fluorescence and bioluminescence techniques, possess the requisite sensitivity and are suitable modalities for studying angiogenesis (61). To this end, various VEGFR ligands have been labeled for PET or SPECT (62–70) and NIR fluorescence imaging (72). Alternative approaches to angiogenesis imaging include the use of VEGFR-targeted monoclonal antibodies (71) and arginine-glycine-aspartic peptides labeled with radioisotopes or NIR dyes, which selectively bind integrins on tumor-associated endothelial cells.
SMART IMAGING AGENTS
An interesting option available to nonnuclear molecular imaging is the use of so-called smart probes, also known as activatable probes. Smart molecular imaging agents can be synthesized such that their signaling properties respond to a variety of relevant tissue-based parameters, including pH, enzyme activity, and metal ion gradients. Of these, preclinical optical molecular imaging agents that become fluorescent on activation by a target enzyme are among the most prevalent (73,74). The development of smart imaging probes for MRI has also been a fruitful area of research. Several prototypical compounds have been reported in which portions of the Gd(III) complex serve as substrates for enzyme activity and the resultant processing of the probe alters the relaxation efficiency of hydrogen protons. The most useful imaging probes demonstrate increased relaxivity on enzymatic processing, and this is usually accomplished by improving water access to the paramagnetic center (75,76). An alternative approach was recently suggested in which probes were prepared such that after specific enzymatic hydrolysis, the agent was able to bind serum proteins, resulting in an in situ macromolecular agent with increased relaxivity (77).
MULTIPLE IMAGING READOUTS: POTENTIAL FOR IMPROVED CHARACTERIZATION
Most notably in the preclinical setting in which optical imaging can be combined with PET or SPECT, it is quite feasible to image more than one molecular event simultaneously (or nearly so). This capability has considerable potential to improve characterization of diseased tissues longitudinally in a single animal. This strategy is also highly useful for modeling complicated clinical dosing regimens in preclinical animal models. In the case of the latter, one could in theory discover, test, and optimize novel therapeutic regimens that affect cellular and molecular processes such as tumor cell proliferation, angiogenesis, and apoptosis. Additionally, during the course of such studies, the most suitable imaging biomarkers can be advanced into the clinic together with the therapeutic regimen if so desired. For example, we recently demonstrated the utility of concomitant use of 3 molecular imaging metrics as potential biomarkers of treatment response to a molecularly targeted therapy used in metastatic colorectal cancer (CRC) (49) and breast cancer (78). Each of the used imaging metrics was selected to assess a unique and important aspect of antitumor therapeutic response. Specifically, we synthesized and validated an optical imaging probe to assess the molecular targeting and tumor cell EGFR occupancy by cetuximab in vivo (NIR800-EGF) and a spectroscopically distinct optical imaging probe to assess the ensuing treatment-induced apoptosis (NIR700-annexin-V). Additionally, changes in tumor proliferation occurring in response to cetuximab were assessed in vivo by 18F-FLT PET. Importantly, the combined noninvasive imaging data illustrate the potential to collect multiple relevant physiologic readouts simultaneously in individual animals. This analysis paradigm revealed that cetuximab, in contrast to its effect on both apoptosis and proliferation in cetuximab-sensitive CRC cells in vitro, induced significant levels of tumor-cell apoptosis but was surprisingly ineffective at reducing tumor-cell proliferation in vivo (Fig. 2) (49). Our in vivo assessment of both cetuximab-sensitive (DiFi) and cetuximab-resistant (HCT-116) cell lines demonstrated that complementary molecular imaging techniques can provide important and accurate information on the biologic effect of a therapy on the tumor. The tight correlation of these imaging measures with direct measurement of EGF binding, proliferation, and apoptosis in the tumor tissue; the ability to detect these changes soon after and throughout dosing; and the ability to obtain this information on a longitudinal, noninvasive basis represent attractive features of molecular imaging.
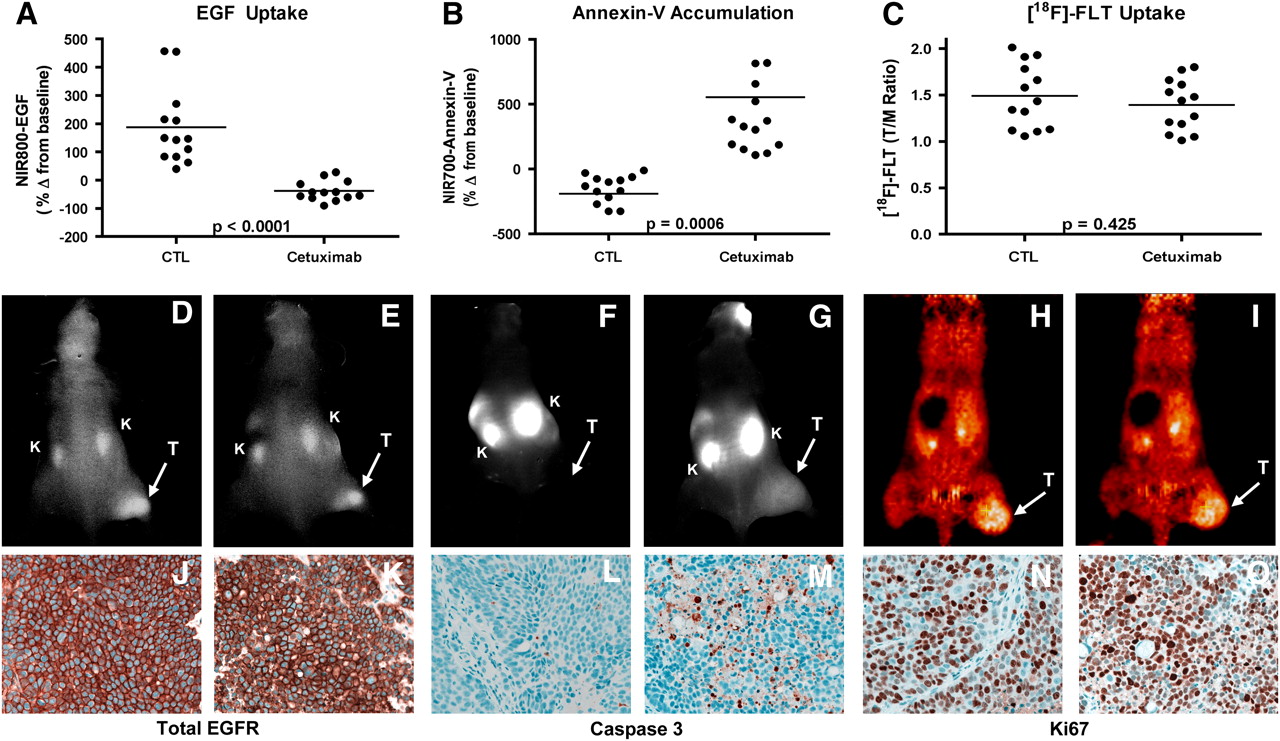
Noninvasive imaging assessment of response to EGFR blockade with cetuximab in CRC xenograft–bearing mice. Treated and untreated cohorts bearing DiFi xenograft tumors were simultaneously imaged with NIR800-EGF, NIR700-annexin-V, and 18F-FLT PET. After cetuximab treatment, mice bearing DiFi tumors, compared with untreated controls (CTL), exhibited significantly reduced NIR800-EGF uptake (A) and increased NIR700-annexin-V uptake (B). No statistical difference in 18F-FLT uptake was observed between treated and untreated mice (C). (D–I) Representative NIR800-EGF, NIR700-annexin-V, and 18F-FLT PET images collected from individual control (D, F, and H) and treated (E, G, and I) mice. Strong agreement between imaging metrics of response and standard immunohistochemistry was observed. Tumors from control (J) and treated (K) animals exhibited similar levels of total EGFR. Treated animals (M), compared with untreated cohorts (L), exhibited elevated caspase 3 staining. No discernible difference in Ki67 staining was observed between tumors from control (N) and treated cohorts (O). T = tumor; K = kidney. (Reprinted with permission of (24).)
Such multiprobe approaches currently can be applied clinically via the sequential scanning of each molecular imaging agent individually. Dual-isotope SPECT offers the potential to conduct such studies simultaneously via the use of radionuclides with γ-ray emissions whose energies can be resolved by the γ-camera. 99mTc and 123I are appealing choices for such dual-isotope studies, although the energy resolution of current NaI-based γ-cameras along with downscatter of 159-keV 123I photons into the 99mTc energy window complicates these measurements. The development of PET/MRI hybrid systems, coupled with continued improvement of MRI molecular imaging agents, may provide another option for true simultaneous readout of multiple molecular imaging probes in humans.
IMPACT OF MOLECULAR IMAGING ON CLINICAL PRACTICE
Although most nuclear medicine departments, regardless of whether they were aware of it, have been engaging in molecular imaging already, the buzz surrounding molecular imaging stems from the paradigm shift that will accompany its full implementation into routine clinical practice. Realization of the full potential of molecular imaging will involve more than simply moving beyond 18F-FDG PET to the incorporation of more PET and SPECT radiotracers. There will be a greater range of purposes served by imaging studies, extending beyond diagnosis to include stratification of patients into the most promising treatment regimen for them individually and early monitoring of response to these personalized therapeutic regimens. The combination of increased specificity of molecular imaging agents and the desire to characterize pathologies to the fullest extent is expected to result in even greater importance for hybrid and multimodality imaging approaches.
It remains to be seen what role nonnuclear methods of molecular imaging will play in clinical practice. PET and SPECT have definite advantages in sensitivity over competing approaches, but imaging modalities that do not involve ionizing radiation may be preferable in scenarios in which multiple scans are performed, such as probing treatment response via comparison of pre- and posttreatment images. Cost, safety, and impact on clinical outcomes will all play a role in the determination of which molecular imaging agents and techniques enter into clinical use. It is also an open question whether the costs associated with the increased scanning implied by the full use of the molecular imaging arsenal will lead to an overall reduction in health care costs through better use of all health care resources.
CONCLUSION
Molecular imaging already is having a profound impact on basic research and drug development that will influence clinical care independent of the translation of these molecular imaging approaches themselves into the clinic. Although molecular imaging appears to provide tools perfectly suited to personalized medicine, there remains some uncertainty as to which molecular imaging agents and techniques may become a standard part of clinical practice. In addition to the scientific aspect of the question, regulatory and reimbursement issues also are important factors influencing the evolution of molecular imaging. SNM has committed itself to “advancing molecular imaging and therapy,” and its recent creation of a Clinical Trials Network is intended to facilitate the incorporation of molecular imaging into the clinical drug trial process. A similar effort may also be needed to provide a framework through which molecular imaging agents themselves might find a path to Food and Drug Administration approval.
The MICoE has created a Web site (www.molecularimagingcenter.org) that provides information and resources for imaging professionals, referring physicians, and patients. The interested reader will find there an extensive bibliography on molecular imaging, including both review articles and the latest research breakthroughs.
Acknowledgments
This work was supported in part by funding from U24 CA126588 (South-Eastern Center for Small-Animal Imaging), 1R01 CA140628, K25 CA127349, and 1P50 CA128323 (Vanderbilt ICMIC Program). The work of Todd E. Peterson, Ph.D., is supported in part by a Career Award at the Scientific Interface from the Burroughs Wellcome Fund.
Footnotes
-
↵* NOTE: FOR CE CREDIT, YOU CAN ACCESS THIS ACTIVITY THROUGH THE SNM WEB SITE (http://www.snm.org/ce_online) THROUGH SEPTEMBER 2011.
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.
- 16.↵
- 17.↵
- 18.↵
- 19.
- 20.↵
- 21.
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.
- 36.
- 37.
- 38.
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- Received for publication January 29, 2009.
- Accepted for publication May 8, 2009.




{kind=link}
{kind=link}